MI
Reading File
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive, well-structured answer. Here is the full response:
Myocardial Infarction (MI)
Definition
MI, commonly called a "heart attack," is necrosis of cardiac muscle resulting from sustained ischemia. The 2018 joint task force of U.S. and European cardiology groups defines it as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia." - Robbins & Kumar Basic Pathology, p. 467
Epidemiology
- ~800,000 MIs per year in the United States (nearly one every 40 seconds)
- ~400,000 deaths annually from MI
- 10% of MIs occur before age 40; 45% before age 65
- Men are at higher risk during middle age; women lose their relative protection after menopause due to falling estrogen and rising inflammatory markers, cholesterol, and blood pressure
- IHD (ischemic heart disease) is the most common cause of death in older women
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
Pathogenesis
Typical Sequence (>90% of cases)
- Plaque disruption - An atheromatous plaque is eroded or ruptured by endothelial injury, intraplaque hemorrhage, or mechanical forces, exposing subendothelial collagen and necrotic plaque contents to circulating blood
- Platelet activation - Platelets adhere, aggregate, and release thromboxane A2, ADP, and serotonin, driving further platelet aggregation and vasospasm
- Coagulation cascade - Tissue factor exposure activates coagulation, adding to the growing thrombus
- Complete occlusion - Within minutes, the enlarging thrombus can completely occlude the coronary artery lumen
Angiography within 4 hours of MI demonstrates thrombosis in ~90% of cases; after 12-24 hours, only 60% - reflecting spontaneous lysis or spasm relaxation in some. This is the basis for early thrombolysis/angioplasty being effective.
Atypical Causes (~10% of cases)
- Vasospasm - with or without atherosclerosis; may be triggered by cocaine, ephedrine, or catecholamines
- Embolism - from left atrial mural thrombus (AF), valve vegetations, prosthetic material, or paradoxical emboli via patent foramen ovale
- Microinfarction - vasculitis, amyloid deposition, sickle cell disease (small intramural vessel pathology)
- Subendocardial ischemia without occlusion - prolonged hypoperfusion/tachycardia on top of fixed coronary stenoses
Robbins, Cotran & Kumar, p. 512
Types of Infarction by Distribution
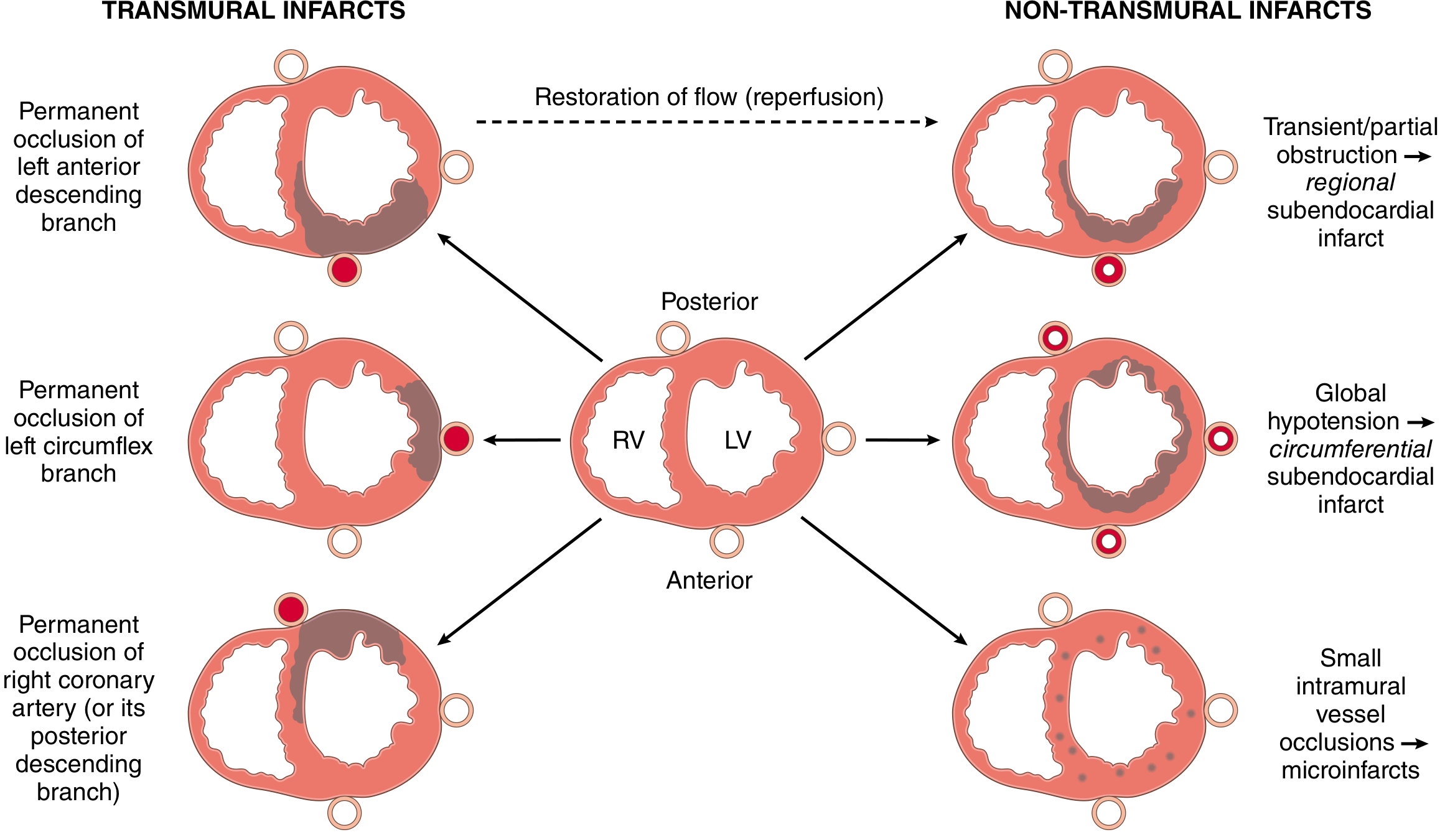
| Type | Artery | Territory |
|---|---|---|
| Transmural | LAD (most common) | Anterior LV wall, anterior septum, apex |
| Transmural | LCX | Lateral LV wall |
| Transmural | RCA | Posterior LV + RV, posterior septum, SA/AV nodes |
| Subendocardial | Any (partial/transient occlusion) | Circumferential inner 1/3; worst with global hypotension |
| Microinfarcts | Small intramural vessels | Scattered foci |
The subendocardium is the most vulnerable zone because coronary vessels are maximally compressed during systole in this region, and it is most distal to the epicardial vessels. - Guyton & Hall, p. 271
Morphological Changes (Timeline)
| Time | Gross Appearance | Light Microscopy | EM / Special |
|---|---|---|---|
| 0-30 min | None | None | Mitochondrial swelling, relaxed myofibrils |
| 30 min - 4 hr | None | Waviness of fibers at border | Membrane defects |
| 4-12 hr | Dark mottling (early) | Pyknosis, vacuolization, early coagulative necrosis begins | - |
| 12-24 hr | Dark mottling | Coagulative necrosis, edema, hemorrhage, early neutrophil infiltration | - |
| 1-3 days | Mottling with yellow-tan softening | Total coagulative necrosis, brisk PMN infiltrate | - |
| 3-7 days | Hyperemic border, central yellow-tan softening | Macrophages appear, phagocytosis of dead cells begins | - |
| 1-3 weeks | Yellow-tan softening, depressed borders | Granulation tissue (fibroblasts + new vessels) replaces necrotic zone | - |
| >2 months | Gray-white scar | Dense collagen scar | Completed healing |
Robbins, Cotran & Kumar - Table 12.5
Key microscopic hallmarks:
- Coagulative necrosis with preservation of cell outlines (ghosts)
- Contraction band necrosis in reperfused infarcts - intense eosinophilic cross bands due to calcium influx causing hypercontracted sarcomeres stuck in tetanus without ATP
- Stunned myocardium - prolonged contractile dysfunction of salvaged (still viable) myocytes that gradually recovers
- Hibernating myocardium - chronically reduced function due to sublethal ischemia, recoverable with revascularization
ECG Changes
Three major membrane polarization abnormalities underlie the ECG picture:
| Defect in Infarcted Cells | Current Flow | ECG Change in Overlying Leads |
|---|---|---|
| Rapid repolarization (K+ channel opening) | Out of infarct | ST elevation |
| Decreased resting membrane potential (K+ loss) | Into infarct during diastole | TQ depression (recorded as ST elevation) |
| Delayed depolarization | Out of infarct | ST elevation |
- Acute phase: ST segment elevation in leads over the infarct; ST depression in reciprocal leads
- Later (days-weeks): ST normalizes; Q waves appear (infarcted muscle is electrically silent and fails to contribute positivity)
- Non-Q-wave infarcts: Less severe but higher risk of subsequent reinfarction
- Ganong's Review of Medical Physiology, p. 534
Localization by leads:
- Anterior MI (LAD): V1-V4
- Lateral MI (LCX): I, aVL, V5-V6
- Inferior MI (RCA): II, III, aVF
- Posterior MI (RCA/LCX): Tall R in V1-V2, ST depression V1-V2 (reciprocal)
Clinical Features
- Chest pain: Prolonged (>30 min), crushing/squeezing/stabbing, often radiates to left arm, jaw, or neck
- Diaphoresis: Profuse sweating
- Nausea/vomiting: Particularly with inferior MI (vagal stimulation)
- Dyspnea: Due to impaired LV contractility and pulmonary congestion
- Rapid, weak pulse
- Silent MI: Up to 25% of cases - especially in diabetics (autonomic neuropathy); discovered only by ECG or biomarkers
- Robbins, Cotran & Kumar, p. 516
Diagnosis
Biomarkers
| Marker | Rise | Peak | Normalization |
|---|---|---|---|
| Troponin I / T | 3-6 hr | 24-48 hr | 7-14 days (gold standard) |
| CK-MB | 4-6 hr | 18-24 hr | 48-72 hr |
| Myoglobin | 1-4 hr | 6-9 hr | 24 hr (earliest, not cardiac-specific) |
| LDH | 24-48 hr | 3-5 days | 7-14 days (late marker) |
Leukocytosis
Elevated WBC occurs in acute MI due to an inflammatory response. Higher WBC counts are associated with poorer short- and long-term prognosis. - Goldman-Cecil Medicine
Complications
Cardiac Causes of Death
-
Decreased cardiac output / Cardiogenic shock
- Occurs when >40% of LV is infarcted
- "Systolic stretch": ischemic/dead muscle bulges outward during systole instead of contracting, dissipating pump energy
- Mortality 40-50% once cardiogenic shock develops
-
Pulmonary edema
- Blood dams back into pulmonary vasculature
- Reduced renal output leads to fluid retention over days, precipitating acute pulmonary edema
-
Ventricular fibrillation
- Most common cause of sudden death
- Two dangerous windows: within 10 minutes, then again at ~1 hour post-infarct
- Mechanisms: K+ loss raises extracellular K+ (increased irritability), injury currents, sympathetic reflexes, and ischemia-lengthened refractory periods creating re-entry
-
Cardiac rupture
- Free wall rupture (hemopericardium/tamponade): days 4-7 during macrophage-mediated softening
- Papillary muscle rupture: acute mitral regurgitation
- Ventricular septal defect: new harsh systolic murmur
Other Complications
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias | Immediate | Most common early complication |
| Pericarditis (Dressler syndrome) | 2-10 days (early); weeks later (late) | Pleuritic chest pain, friction rub |
| LV thrombus | Days-weeks | Embolic stroke risk |
| Aneurysm formation | Weeks-months | Persistent ST elevation, bulging scar |
| Heart failure | Acute or chronic | Proportional to infarct size |
Guyton & Hall, pp. 271-273
Management Overview
Acute Phase (STEMI)
- Aspirin + P2Y12 inhibitor (clopidogrel, ticagrelor, or prasugrel) - dual antiplatelet therapy
- Anticoagulation (heparin / LMWH / bivalirudin)
- Primary PCI (percutaneous coronary intervention) - preferred reperfusion if available within 90 min
- Fibrinolysis - if PCI not available within 120 min of first medical contact
- Nitrates - pain relief, preload reduction
- Morphine - analgesia (though evidence now suggests caution due to possible platelet interaction)
- Beta-blockers - reduce myocardial oxygen demand, antiarrhythmic
- Oxygen - only if SpO2 <90%
Secondary Prevention
- Beta-blockers: Traditionally given post-MI; a 2025 meta-analysis found that in patients without reduced EF or heart failure, the benefit of long-term beta-blockers is uncertain - this challenges conventional practice
- ACE inhibitors/ARBs: Reduce remodeling, especially with reduced EF
- High-intensity statins: Plaque stabilization and LDL lowering
- Aldosterone antagonists (eplerenone/spironolactone): In patients with EF <40% + HF or diabetes
- ICD: If EF remains <35% after 40+ days
Recent Evidence Update
- Beta-blockers post-MI without reduced EF (2025 meta-analysis, PMID 39298680): Challenges routine long-term beta-blocker use in preserved EF post-MI patients
- Sudden cardiac death post-MI (2024, PMID 39378245): Individual participant data from pooled cohorts characterizing SCD risk stratification after MI
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.), pp. 511-516
- Robbins & Kumar Basic Pathology, pp. 467-471
- Ganong's Review of Medical Physiology (26th ed.), pp. 534-536
- Guyton & Hall Textbook of Medical Physiology, pp. 271-273
- Braunwald's Heart Disease (12th ed.)
This is a shared conversation. Sign in to Orris to start your own chat.