Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed for a thorough, well-sourced answer. Here is a full explanation of ARDS mechanism:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition and Diagnostic Criteria (Berlin Definition)
ARDS is an inflammatory lung injury characterized by bilateral infiltrates on chest radiograph or CT, a PaO2/FiO2 (P/F) ratio <300 mmHg on at least 5 cm H2O PEEP, onset within 7 days of a recognized insult, and pulmonary edema not fully explained by cardiac failure or fluid overload. Severity is stratified as mild (P/F 200-300), moderate (P/F 100-200), and severe (P/F <100).
- Sabiston Textbook of Surgery, p. 940
Inciting Events: Direct vs. Indirect Injury
Risk factors divide into two broad categories:
| Direct (Pulmonary) Injury | Indirect (Nonpulmonary) Injury |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis |
| Aspiration of gastric contents | Major trauma |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic inhalation / near-drowning | Pancreatitis |
| Reperfusion injury (post-lung transplant) | Cardiopulmonary bypass, drug overdose |
Sepsis carries the highest risk (up to 43% of cases develop ARDS). The presence of multiple risk factors compounds the likelihood significantly.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3145
Core Pathophysiology: Breakdown of the Alveolar-Capillary Barrier
The central event in ARDS is increased permeability of the alveolar-capillary membrane, distinguishing it from cardiogenic (hydrostatic) pulmonary edema. The alveoli fill with protein-rich, exudative fluid rather than transudate. This triggers a cascade:
- Alveolar flooding reduces functional residual capacity (FRC)
- Right-to-left shunting and low V/Q regions cause profound hypoxemia refractory to supplemental O2
- Dead space ventilation increases significantly (alveoli are ventilated but not perfused)
- Lung compliance falls, requiring higher airway pressures to deliver tidal volumes
- Pulmonary hypertension develops via hypoxic vasoconstriction, intravascular fibrin deposition, and compression of vessels by positive-pressure ventilation
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3144-3145
Pathological Phases: Diffuse Alveolar Damage (DAD)
ARDS progresses through three overlapping stages:
1. Exudative Phase (Days 1-7)
- Hyaline membranes form (composed of cellular debris, plasma proteins, and surfactant components)
- Protein-rich fluid floods alveolar spaces
- Widespread epithelial disruption (particularly Type I pneumocytes)
- Heavy neutrophil infiltration of interstitium and airspaces
2. Proliferative Phase (Days 7-14)
- Hyaline membranes undergo organization
- Early fibrosis appears
- Pulmonary capillary obliteration begins
- Interstitial and alveolar collagen deposition
- Neutrophil numbers decline; Type II pneumocyte proliferation attempts repair
3. Fibrotic Phase (>14 days, in a subset)
-
Established pulmonary fibrosis
-
Notably, elevated N-terminal procollagen peptide III (a marker of collagen synthesis) can be detected in BAL fluid as early as 24 hours after onset - suggesting fibroproliferation begins simultaneously with acute inflammation, not after it
-
Murray & Nadel's Textbook of Respiratory Medicine, p. 3144-3145
Cellular and Molecular Mechanisms
Alveolar Epithelial Injury (Key Initiating Event)
Damage to the alveolar epithelium - especially Type I pneumocytes (covering ~95% of the alveolar surface) - is considered the pivotal precipitating event. Multiple mechanisms drive epithelial cell death:
- Direct cytotoxicity from pathogens or toxins
- Neutrophil-derived oxidants and proteases
- Fas/FasL-mediated apoptosis
- Endoplasmic reticulum stress
Loss of Type I cells impairs the epithelial barrier, allowing protein-rich fluid to pour into the airspace. Loss of Type II pneumocytes reduces surfactant production, worsening alveolar collapse and increasing surface tension.
Neutrophil-Mediated Injury
Activated neutrophils are the central effector cells. Sequestration of neutrophils within alveolar and interstitial spaces is driven by:
- TNF-alpha and IL-8 (released from macrophages and epithelial cells)
- Complement activation (C5a)
- Platelet-activating factor (PAF)
Once activated, neutrophils release:
- Proteases (elastase, matrix metalloproteinases) - degrade the extracellular matrix and epithelial tight junctions
- Reactive oxygen species (ROS) - oxidize lipid membranes and proteins, further disrupting barrier integrity
- Leukotrienes and prostaglandins - amplify local inflammation and increase vascular permeability
Microvascular Endothelial Injury
The pulmonary microvascular endothelium is simultaneously damaged:
- Cytokines cause endothelial cell retraction and gap formation
- Loss of endothelial barrier allows plasma proteins and fluid to leak into the interstitium and airspaces
- Endothelial activation upregulates adhesion molecules (ICAM-1, E-selectin), recruiting more neutrophils
Inflammation-Coagulation Crosstalk
Inflammation and coagulation are deeply interconnected in ARDS:
-
TNF-alpha increases thrombin/fibrin formation, stimulates tissue factor expression on endothelial cells, and inhibits fibrinolysis
-
Fibrin fragments are chemotactic for neutrophils, amplifying the inflammatory loop
-
Intravascular fibrin deposition occludes pulmonary microvessels, contributing to pulmonary hypertension and dead space
-
Endogenous activated protein C (an anticoagulant) has anti-inflammatory effects including downregulating IL-6 and attenuating neutrophil activation; its function is impaired in ARDS
-
Murray & Nadel's Textbook of Respiratory Medicine, p. 3163-3164
Surfactant Dysfunction
Surfactant is both depleted (Type II pneumocyte loss) and inactivated (by plasma proteins leaking into alveoli). In pancreatitis-associated ARDS, phospholipase A2 directly degrades surfactant enzymatically. Loss of surfactant increases alveolar surface tension, causing diffuse microatelectasis and further worsening shunt.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 2961
Impaired Alveolar Fluid Clearance
Normal alveolar fluid reabsorption depends on apical Na+ channels (ENaC) on Type II pneumocytes and basolateral Na+/K+ ATPases. In ARDS:
-
Hypoxia downregulates ENaC expression and activity
-
Hypoxia inhibits Na+/K+ ATPase activity
-
Increased nitric oxide in the lung impairs the catecholamine-driven upregulation of fluid reabsorption
-
The net result is flooded alveoli that cannot clear edema fluid effectively
-
Murray & Nadel's Textbook of Respiratory Medicine, p. 3201-3203
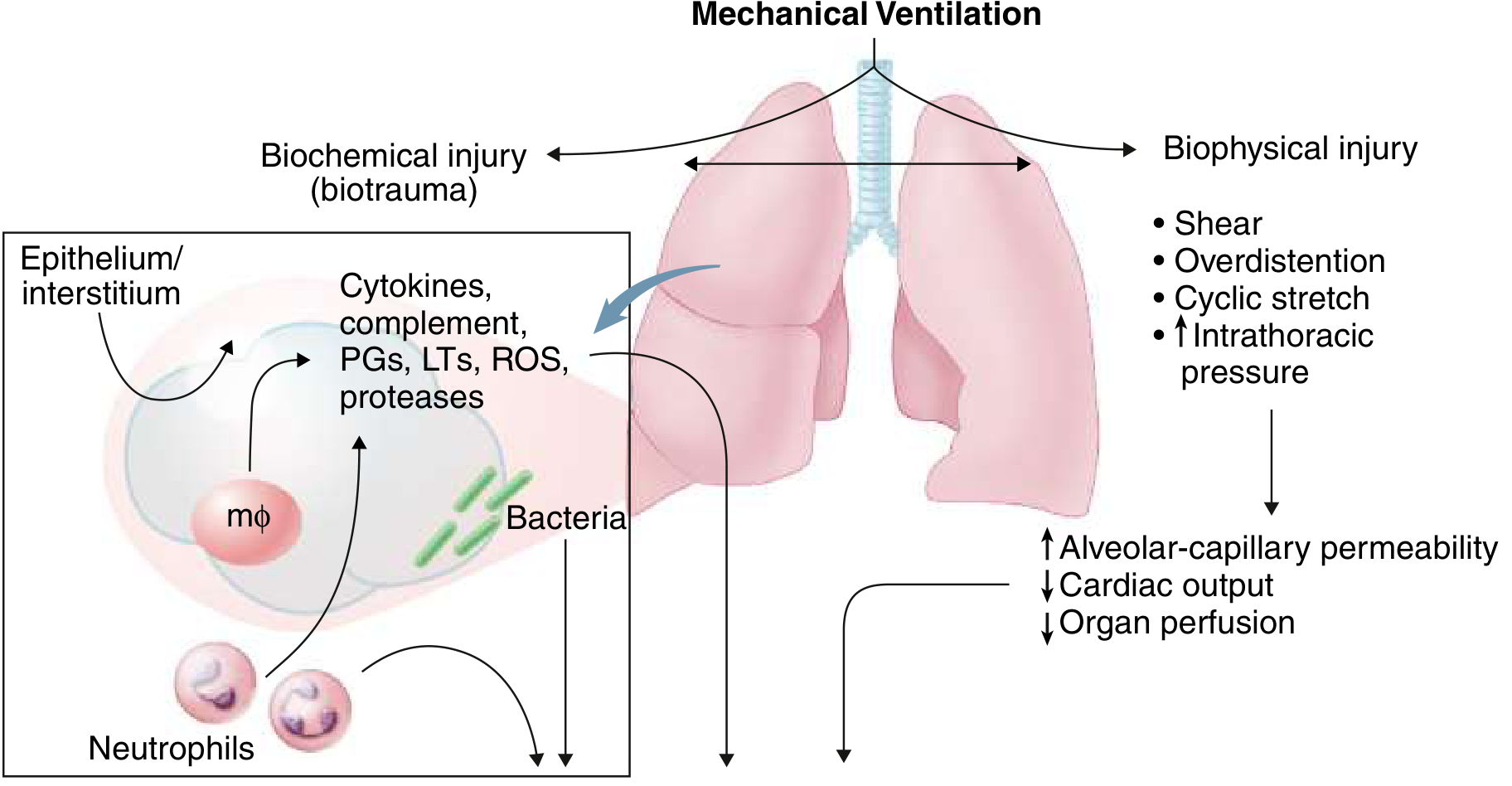
Ventilator-Induced Lung Injury (VILI): Amplifying the Damage
Mechanical ventilation, while life-saving, can worsen ARDS through two mechanisms:
Biophysical injury:
- Volutrauma/barotrauma: Overdistention of relatively normal alveoli adjacent to collapsed regions
- Atelectrauma: Cyclic opening and closing (shear stress) of unstable alveolar units
Biochemical injury (biotrauma):
- Injurious ventilation promotes release of cytokines, complement, ROS, and proteases from macrophages and neutrophils within the lung
- These mediators spill into the systemic circulation and can contribute to multi-organ failure
This is why lung-protective ventilation (tidal volumes 6-8 mL/kg predicted body weight, plateau pressure ≤30 cm H2O) is the cornerstone of ARDS management.
- Goldman-Cecil Medicine, p. 1066-1067
Summary: The Unified Mechanism
Inciting Insult (sepsis, pneumonia, aspiration, trauma, etc.)
↓
Macrophage/epithelial cell activation
↓
Release of TNF-α, IL-1β, IL-8, complement, PAF
↓
Neutrophil sequestration in alveolar-capillary bed
↓
Neutrophil release of ROS, proteases, leukotrienes
↓
Alveolar epithelial + microvascular endothelial injury
↓
Increased alveolar-capillary permeability
↓
Protein-rich exudate floods alveoli
↓
Surfactant dysfunction + impaired Na+ fluid clearance
↓
Alveolar collapse, V/Q mismatch, shunt → hypoxemia
↓
↓ Compliance, ↑ dead space, pulmonary hypertension
↓
Diffuse Alveolar Damage (DAD) + risk of fibroproliferation
Mechanical ventilation can perpetuate the cycle through biotrauma, releasing inflammatory mediators that worsen both lung injury and distal organ dysfunction.
Key sources:
- Murray & Nadel's Textbook of Respiratory Medicine (2-Volume)
- Goldman-Cecil Medicine
- Sabiston Textbook of Surgery
This is a shared conversation. Sign in to Orris to start your own chat.