Explain me urea cycle
Reading File
Finding Sources
Searching Images
urea cycle diagram biochemistry
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Reading File
Here is a comprehensive explanation of the urea cycle, synthesized from Basic Medical Biochemistry: A Clinical Approach (6e) and Biochemistry, 8th ed (Lippincott Illustrated Reviews).
The Urea Cycle
Overview & Purpose
The urea cycle (also called the Krebs–Henseleit cycle, proposed in 1932) is the primary pathway by which the body disposes of ammonia (NH₃/NH₄⁺), a toxic byproduct of amino acid catabolism. Ammonia is particularly toxic to the brain and CNS. The liver converts it to the non-toxic, water-soluble compound urea, which travels in the blood to the kidneys for urinary excretion.
- Normal blood ammonia: 30–60 μM
- Urea accounts for ~90% of urinary nitrogen
- The cycle is almost exclusively hepatic
Compartmentalization
The cycle spans two cellular compartments:
| Location | Reactions |
|---|---|
| Mitochondrial matrix | Steps 1 & 2 (carbamoyl phosphate & citrulline synthesis) |
| Cytosol | Steps 3, 4 & 5 (argininosuccinate → arginine → urea) |
An antiporter on the inner mitochondrial membrane exchanges ornithine (entering) for citrulline (exiting).
The 5 Reactions
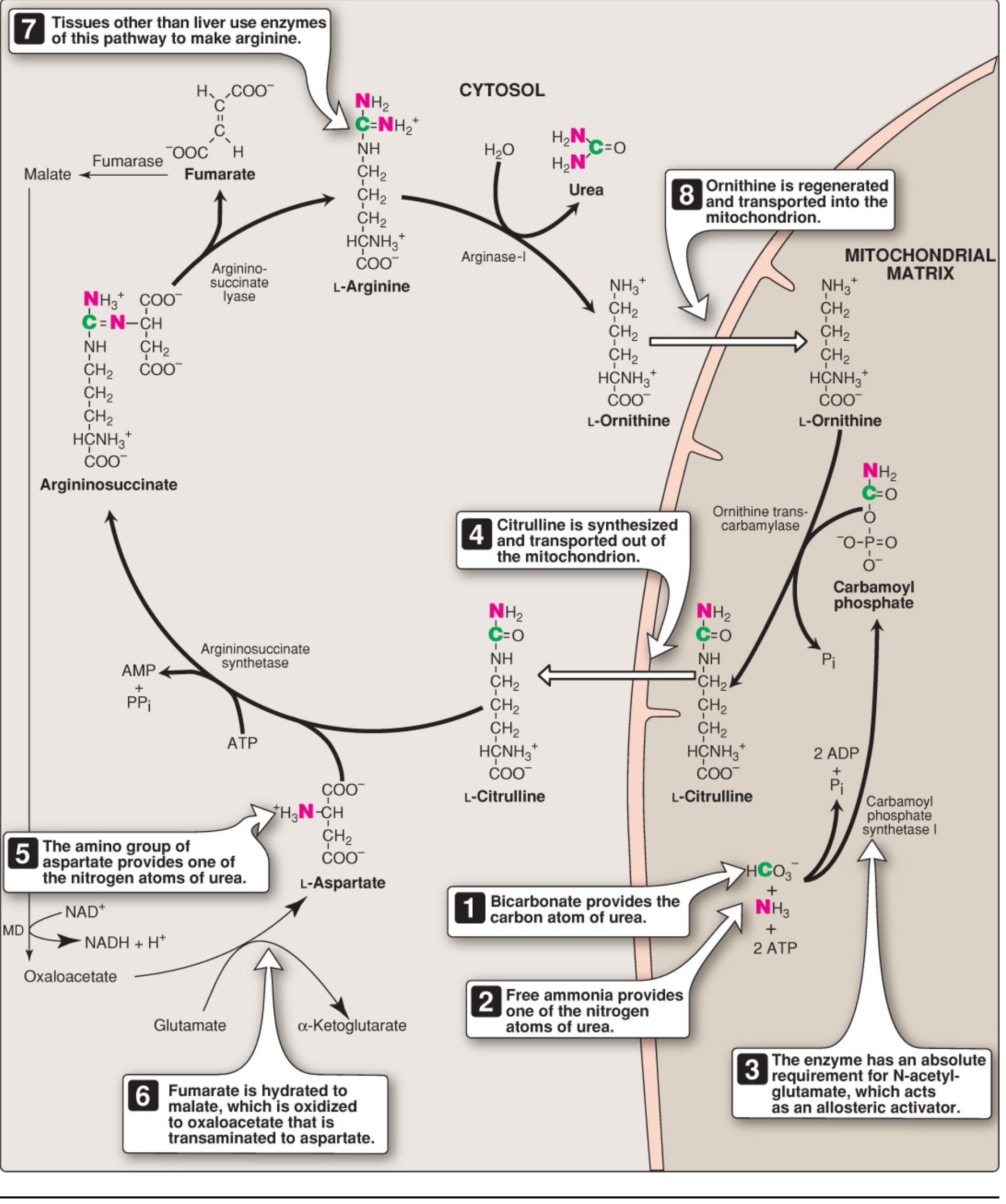
Step 1 — Carbamoyl Phosphate Synthesis (Mitochondria)
NH₄⁺ + HCO₃⁻ + 2 ATP → Carbamoyl phosphate + 2 ADP + Pi
- Enzyme: Carbamoyl Phosphate Synthetase I (CPS I)
- Location: Mitochondrial matrix
- Requires cleavage of 2 ATP — energetically costly
- Absolute requirement for the allosteric activator N-acetylglutamate (NAG)
- This is the committed, rate-limiting step of the cycle
- (CPS II, in the cytosol, uses glutamine as N-source for pyrimidine synthesis — a separate enzyme)
Step 2 — Citrulline Synthesis (Mitochondria)
Carbamoyl phosphate + Ornithine → Citrulline + Pi
- Enzyme: Ornithine Transcarbamylase (OTC)
- Ornithine is the regenerating carrier molecule (analogous to oxaloacetate in the TCA cycle)
- Citrulline exits to the cytosol via the ornithine/citrulline antiporter
Step 3 — Argininosuccinate Synthesis (Cytosol)
Citrulline + Aspartate + ATP → Argininosuccinate + AMP + PPi
- Enzyme: Argininosuccinate Synthetase (ASS)
- Aspartate donates the second nitrogen atom of urea
- Cleavage of ATP → AMP + PPi (equivalent to 2 ATP used)
- Aspartate is generated by transamination of oxaloacetate by AST
Step 4 — Argininosuccinate Cleavage (Cytosol)
Argininosuccinate → Arginine + Fumarate
- Enzyme: Argininosuccinate Lyase (ASL)
- Fumarate is a key byproduct — it links the urea cycle with the TCA cycle (the "Krebs bi-cycle")
- Fumarate → malate → oxaloacetate → transaminated back to aspartate (recycling the nitrogen carrier)
Step 5 — Arginine Hydrolysis / Urea Release (Cytosol)
Arginine + H₂O → Urea + Ornithine
- Enzyme: Arginase I (virtually exclusive to the liver)
- Releases urea from arginine's guanidinium group
- Regenerates ornithine, which re-enters the mitochondria to restart the cycle
Overall Stoichiometry
Aspartate + NH₃ + HCO₃⁻ + 3 ATP + H₂O
→ Urea + Fumarate + 2 ADP + AMP + 2 Pi + PPi
- 4 high-energy phosphate bonds are consumed per urea molecule
- The reaction is irreversible (large negative ΔG)
- Both nitrogen atoms of urea ultimately come from glutamate: one via oxidative deamination (→ NH₃) and one via transamination of oxaloacetate (→ aspartate)
Regulation
| Mechanism | Detail |
|---|---|
| Substrate availability (short-term) | More NH₃ → faster cycle (feed-forward regulation) |
| N-acetylglutamate (NAG) — allosteric | Activates CPS I; synthesized by NAG synthase from acetyl-CoA + glutamate; arginine stimulates NAG synthesis |
| Enzyme induction (long-term) | High-protein diet or prolonged fasting upregulates all urea cycle enzymes |
Nitrogen Sources Summary
| Nitrogen Atom | Source | Entry Point |
|---|---|---|
| N₁ (from NH₃) | Glutamate → oxidative deamination (GDH) | Carbamoyl phosphate (Step 1) |
| N₂ (from aspartate) | Glutamate → transamination of OAA (AST) | Step 3 |
| Carbon of urea | CO₂ (as HCO₃⁻) | Step 1 |
Link to TCA Cycle — The "Krebs Bi-Cycle"
The fumarate released in Step 4 feeds directly into the TCA cycle pathway:
Fumarate → Malate → Oxaloacetate → Aspartate (recycled back to the urea cycle)
This bidirectional connection means amino acid nitrogen flows continuously into urea via the coordinated action of both cycles.
Clinical Relevance
| Defect | Effect |
|---|---|
| CPS I deficiency | Hyperammonemia; elevated NH₃ |
| OTC deficiency (X-linked, most common) | Hyperammonemia + orotic aciduria (carbamoyl phosphate floods pyrimidine synthesis) |
| ASS deficiency (Citrullinemia type I) | Elevated citrulline |
| ASL deficiency (Argininosuccinic aciduria) | Elevated argininosuccinate |
| Arginase deficiency | Hyperargininemia; spastic diplegia |
| Liver failure | Reduced urea cycle capacity → hepatic encephalopathy from NH₃ accumulation |
Sources: Basic Medical Biochemistry: A Clinical Approach, 6e, pp. 1321–1326 | Biochemistry, 8th ed (Lippincott Illustrated Reviews), pp. 712–715
Explain me structure of protein and it’s functions
Finding Sources
Searching Images
protein structure primary secondary tertiary quaternary
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Now I have comprehensive material to deliver a full explanation. Let me compile it.
Structure of Proteins
Proteins are the most structurally and functionally diverse macromolecules in the body. Their complexity is analyzed at four organizational levels, each building upon the last.
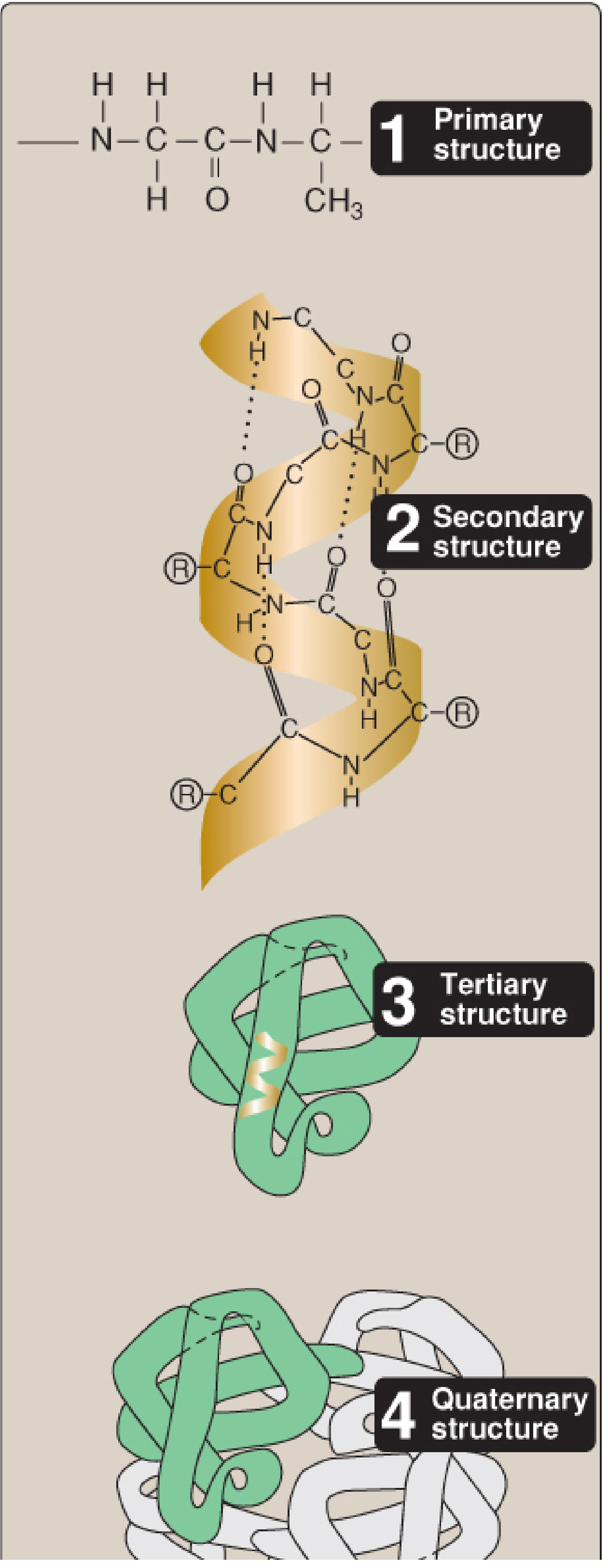
The Building Blocks — Amino Acids & Peptide Bonds
Proteins are linear chains of amino acids joined by peptide bonds — amide linkages between the α-carboxyl group of one amino acid and the α-amino group of the next.
Key features of the peptide bond:
- Has partial double-bond character → it is rigid and planar (no free rotation around it)
- Is uncharged at physiological pH (the C=O and N-H groups are polar but not ionizable)
- The N-terminal (free amino end) is written to the left; the C-terminal (free carboxyl end) to the right
- A chain of ≥50 amino acids is called a polypeptide
- Average molecular weight of one amino acid: ~120 Da
Level 1 — Primary Structure
The linear sequence of amino acids in the polypeptide chain. This sequence is:
- Encoded directly by DNA
- The ultimate determinant of all higher-order structure and function
- The basis for protein identity — mutations here (e.g., sickle-cell disease) cause misfolding and disease
Level 2 — Secondary Structure
Regular, repetitive three-dimensional conformations formed by local regions of the backbone, stabilized exclusively by backbone hydrogen bonds (not side chains).
α-Helix
- A right-handed spiral with the side chains (R groups) pointing outward
- Stabilized by H-bonds: the C=O of each peptide bond to the N-H four residues ahead
- 3.6 amino acids per turn
- Proline disrupts the helix (its ring creates a rigid kink)
- Examples: keratin (hair, skin), large portions of myoglobin
β-Sheet (β-Pleated Sheet)
- Two or more fully extended β-strands aligned laterally, linked by perpendicular H-bonds between strands
- Strands can be antiparallel (N-termini alternate) or parallel (N-termini on same side)
- R groups extend alternately above and below the plane of the sheet
- Has a slight right-handed twist
β-Bends (β-Turns / Reverse Turns)
- 4-amino acid loops that reverse the direction of the polypeptide chain
- Allow the protein to form a compact, globular shape
- Often contain proline and glycine
- Stabilized by H-bonds between residue 1 and residue 4 of the turn
- Frequently located on the protein surface
Supersecondary Structures (Motifs)
Secondary elements combine into geometric patterns:
- β-α-β motif (β strand, helix, β strand)
- β-meander (antiparallel β-strands connected by loops)
- β-barrel (β-strands folded into a barrel shape)
- Helix-loop-helix (found in DNA-binding transcription factors)
Level 3 — Tertiary Structure
The overall three-dimensional folding of a single polypeptide chain. It encompasses:
- Arrangement of secondary elements and loops into domains (functional/structural units, usually >200 residues have ≥2 domains)
- Compact globular structure with hydrophobic core and hydrophilic surface
Four Stabilizing Interactions
| Interaction | Type | Description |
|---|---|---|
| Disulfide bonds (–S–S–) | Covalent | Between two cysteine residues; resist denaturation; abundant in secreted proteins (e.g., immunoglobulins) |
| Hydrophobic interactions | Non-covalent | Nonpolar side chains cluster in the interior, away from water — the main driving force of folding |
| Hydrogen bonds | Non-covalent | Between polar side chains (e.g., –OH of Ser/Thr) and electron-rich atoms |
| Ionic interactions (salt bridges) | Non-covalent | Between oppositely charged groups (e.g., –COO⁻ of Asp/Glu with –NH₃⁺ of Lys) |
Protein Folding & Chaperones
- Folding occurs within seconds to minutes via ordered, nonrandom pathways
- Driven by the hydrophobic effect: hydrophobic groups come together as water is expelled
- Molecular chaperones (Heat-Shock Proteins, HSPs) facilitate correct folding:
- Hsp70: binds extended nascent chains, prevents premature folding
- Hsp60: cage-like barrel where misfolded proteins refold in an isolated environment
- Without chaperones, many proteins misfold and aggregate
Protein Denaturation
Disruption of secondary/tertiary structure without breaking peptide bonds, caused by:
heat, urea, strong acids/bases, organic solvents, heavy metals, detergents.
Most denatured proteins do not spontaneously refold and precipitate out of solution.
Level 4 — Quaternary Structure
The arrangement of two or more polypeptide subunits into one functional protein complex.
- Subunits held together by non-covalent interactions (H-bonds, ionic, hydrophobic)
- Subunits may be identical (homomeric) or different (heteromeric)
- Classic example: Hemoglobin — a tetramer (2α + 2β subunits) exhibiting cooperative O₂ binding
- Isoforms / Isoenzymes: proteins performing the same function but with different primary structures (tissue-specific expression)
Protein Misfolding & Disease
When proteins fold incorrectly, β-sheet-rich aggregates called amyloid can form:
| Disease | Protein involved |
|---|---|
| Alzheimer disease | Amyloid-β peptide (plaques) + hyperphosphorylated tau (tangles) |
| Parkinson disease | α-Synuclein |
| Prion diseases (CJD, scrapie) | PrP^C → PrP^Sc (α-helices → β-sheets; infectious conformational change) |
Functions of Proteins
| Function | Examples |
|---|---|
| Structural | Collagen (connective tissue), keratin (hair/nails), elastin |
| Enzymatic | All metabolic catalysis (amylase, kinases, polymerases) |
| Transport | Hemoglobin (O₂), albumin (fatty acids, drugs), transferrin (iron) |
| Signaling / Hormonal | Insulin, growth hormone, cytokines |
| Receptor | G-protein coupled receptors, ion channel receptors |
| Immune defense | Antibodies (immunoglobulins), complement proteins |
| Contractile/Motor | Actin, myosin (muscle contraction), dynein, kinesin |
| Storage | Ferritin (iron), casein (milk protein, amino acid store) |
| Regulatory | Transcription factors, histones (gene regulation) |
| Protective/Clotting | Fibrinogen, thrombin, clotting factors |
Sources: Biochemistry, 8th ed (Lippincott Illustrated Reviews), pp. 51–83 | Henry's Clinical Diagnosis and Management by Laboratory Methods, pp. 321–323
Explain this pdf for a seminar
Reading File
You've run out of free credits. Sign up for a free account to continue using Orris.
This is a shared conversation. Sign in to Orris to start your own chat.