Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive, high-quality content from Murray & Nadel's Textbook of Respiratory Medicine. I have enough to write a thorough, well-organized answer. Let me compose it now.
Acute Respiratory Distress Syndrome (ARDS) - Mechanism
ARDS is a syndrome of acute, non-cardiogenic pulmonary edema caused by diffuse alveolar-capillary injury. The core problem is a breakdown of the alveolar-capillary barrier, which floods the alveoli with protein-rich exudate and triggers a cascade of inflammation, structural damage, and impaired gas exchange.
Triggers: Direct vs. Indirect Injury
The initial insult reaches the lung by one of two routes:
| Direct (Pulmonary) | Indirect (Nonpulmonary) |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis (most common overall cause, ~30% risk) |
| Aspiration of gastric contents | Major trauma |
| Pulmonary contusion | Pancreatitis |
| Toxic inhalation, near-drowning | Multiple blood transfusions |
| Reperfusion injury (post-transplant) | Drug overdose, cardiopulmonary bypass |
Sepsis is the single most common precipitant. Alcoholism doubles the risk, likely through impaired alveolar epithelial function.
- Murray & Nadel's Textbook of Respiratory Medicine, Table 134.2
Core Mechanism: Alveolar-Capillary Barrier Failure
Normally, the alveolar-capillary membrane is a tight barrier consisting of:
- The pulmonary microvascular endothelium (inner surface)
- The alveolar epithelium - Type I pneumocytes (covering ~95% of the surface) and Type II pneumocytes (surfactant production, barrier repair)
In ARDS, both layers are damaged:
Endothelial injury - The initial insult activates endothelial cells, disrupting tight junctions and adherens junctions (which rely on VE-cadherin). This increases vascular permeability and allows fluid and proteins to leak into the interstitium.
Epithelial injury - Type I pneumocyte death impairs the barrier. Type II pneumocyte injury reduces surfactant production and impairs alveolar fluid clearance (normally driven by sodium transporters that actively pump fluid out of the airspace). With both arms of active fluid clearance disabled, protein-rich edema floods the alveolar spaces.
The result is diffuse alveolar damage (DAD) - the histologic hallmark of ARDS, seen in approximately 50% of ARDS patients at autopsy or biopsy.
- Murray & Nadel's Textbook of Respiratory Medicine, pp. 3145-3147
The Three Pathologic Phases
1. Exudative Phase (Days 1-7)
- Hyaline membranes form in alveoli, composed of cellular debris, proteins, and surfactant fragments
- Protein-rich fluid fills the alveolar spaces (non-hydrostatic, exudative edema)
- Widespread Type I and Type II pneumocyte death
- Massive neutrophil infiltration of the interstitium and airspaces
- Surfactant is inactivated by plasma proteins leaking into the alveoli, and its production decreases due to Type II cell injury - this promotes alveolar collapse
2. Fibroproliferative Phase (Days 7-21)
- Hyaline membranes are reorganized
- Fibrosis begins to appear
- Obliteration of pulmonary capillaries
- Interstitial and alveolar collagen deposition
- Neutrophil numbers decrease; the edema partially resolves
- Importantly, markers of collagen synthesis (N-terminal procollagen peptide III) can be detected in BAL fluid as early as 24 hours after onset, suggesting fibroproliferation may begin simultaneously with inflammation rather than strictly after
3. Fibrotic Phase (>2 weeks)
- A subset of patients develop established pulmonary fibrosis
- This is associated with prolonged ventilator dependence and worsened prognosis
- Murray & Nadel's, pp. 3074-3076
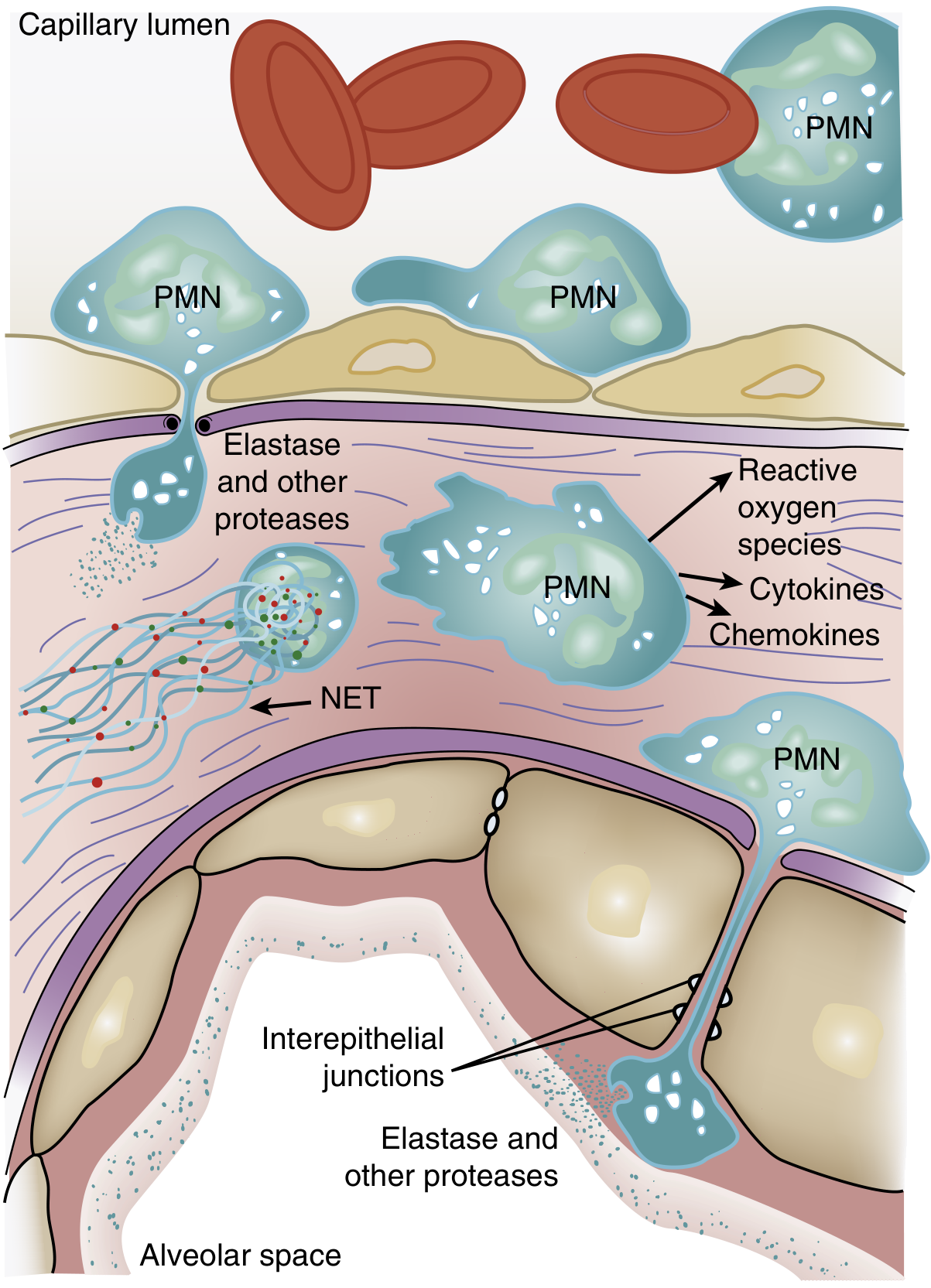
Inflammatory Cascade: Neutrophils as Central Mediators
One of the earliest signs of ARDS - even before frank hypoxemia - is transient leukopenia, caused by sequestration of neutrophils in the pulmonary microvasculature. Here is why:
- Pulmonary capillaries are narrower than neutrophils, so neutrophils must deform to pass through
- Activated neutrophils become "stiff" (via actin cytoskeleton changes) and cannot deform, so they become trapped in capillaries
- Sequestered neutrophils induce endothelial barrier breakdown, facilitating their transmigration into the lung interstitium and alveolar space
Once in the lung, activated neutrophils release a broad arsenal of damaging agents:
| Agent | Effect |
|---|---|
| Reactive oxygen species (ROS) | Oxidative damage to cell membranes, proteins |
| Neutrophil elastase (NE) | Degrades epithelial/endothelial cadherins, destroying adherens junctions; degrades growth factors |
| Metalloproteinases | Matrix destruction |
| TNF-α, IL-1β | Amplify the inflammatory response, recruit more neutrophils |
| Neutrophil extracellular traps (NETs) | Web-like DNA/histone/antimicrobial peptide structures that damage endothelium and promote thrombosis |
Key Signaling Pathways
- p38 MAPK - Activated by LPS, stimulates TNF-α production and macrophage inflammatory protein-2 (a neutrophil chemokine); inhibition attenuates neutrophil migration into alveoli in animal models
- PI3K-γ - Activated in neutrophils by IL-8 and bacterial peptides; knockout mice show decreased neutrophil accumulation and lung injury
- Renin-angiotensin system - ACE knockout mice have improved outcomes in experimental lung injury, implicating angiotensin II in ARDS pathogenesis
- Murray & Nadel's, pp. 3126-3149
Macrophage Contributions
Alveolar macrophages are also activated early and release:
- TNF-α and IL-1β - primary pro-inflammatory cytokines
- IL-8 - a potent neutrophil chemokine
- These amplify neutrophil recruitment in a self-reinforcing loop
Surfactant Dysfunction
- Plasma proteins leaking into alveoli inhibit surfactant function
- Type II pneumocyte injury reduces surfactant synthesis
- The net result is increased alveolar surface tension, promoting collapse of small airways and alveoli
- This contributes to the ventilation-perfusion (V/Q) mismatch and intrapulmonary shunt seen clinically
Unlike neonatal RDS (where surfactant deficiency is the primary driver), exogenous surfactant replacement trials in adult ARDS have not shown mortality benefit, suggesting surfactant dysfunction is one of several parallel mechanisms rather than the central driver.
- Murray & Nadel's, pp. 3101-3105
Physiological Consequences
All of the above produce the characteristic physiology of ARDS:
- Profound hypoxemia - Alveolar flooding causes V/Q mismatch and right-to-left shunting; this is refractory to supplemental oxygen alone because the shunted blood bypasses ventilated alveoli entirely
- Reduced lung compliance - Fluid-filled, collapsed alveoli are stiff; the lung behaves like a "sponge," with gravity-dependent consolidation
- Increased dead-space ventilation - Despite elevated minute ventilation, CO2 clearance is impaired; increased dead-space fraction is associated with higher mortality
- Pulmonary hypertension - From hypoxic vasoconstriction, intravascular fibrin deposition, and vascular compression by positive-pressure ventilation
Berlin Definition Severity (for context)
| Severity | PaO2/FiO2 ratio | PEEP requirement |
|---|---|---|
| Mild | 200-300 mmHg | ≥5 cm H2O |
| Moderate | 100-200 mmHg | ≥5 cm H2O |
| Severe | <100 mmHg | ≥5 cm H2O |
Summary
ARDS begins with an insult (direct or indirect) that activates the innate immune system. The resulting cytokine storm drives neutrophil sequestration in pulmonary capillaries, transmigration into the lung, and release of proteases, ROS, and NETs. This destroys the alveolar-capillary barrier, causing protein-rich exudate to flood the alveoli (DAD). Surfactant dysfunction, alveolar collapse, and capillary obliteration follow. The lung progresses through exudative, fibroproliferative, and fibrotic phases - with mortality ranging from ~35% in mild ARDS to ~46% in severe disease.
Sources: Murray & Nadel's Textbook of Respiratory Medicine (Ch. 134); Current Surgical Therapy 14e; Barash Clinical Anesthesia 9e
This is a shared conversation. Sign in to Orris to start your own chat.