Give an long easy about the apoptosis and as well as necrosis and this differences
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
I now have comprehensive, authoritative content from Robbins & Kumar Basic Pathology and Robbins, Cotran & Kumar Pathologic Basis of Disease. I have enough to write a thorough, detailed essay. Let me compose it now.
Apoptosis and Necrosis: A Comprehensive Essay on Cell Death
Introduction
Cell death is one of the most fundamental processes in biology and pathology. Every multicellular organism depends on a precise balance between cell survival, proliferation, and death. When this balance is disrupted, diseases ranging from cancer to neurodegeneration can result. Two major forms of cell death have been identified and extensively studied: apoptosis and necrosis. Although both result in the loss of a cell, they differ profoundly in their triggers, mechanisms, morphology, biochemical signatures, and consequences for surrounding tissue. Understanding them is not merely academic - it underpins modern approaches to treating cancer, ischemic injury, infection, and inflammation.
Part I: Necrosis
Definition and General Concept
Necrosis is the form of cell death that occurs when cells are subjected to severe, overwhelming injury that they cannot survive or manage in an orderly way. It is inherently a pathologic process - it does not serve a physiological purpose in the body. The word "necrosis" comes from the Greek nekros, meaning "dead body," and that etymology captures its essence: necrosis is death imposed from the outside, not orchestrated from within.
As stated in Robbins, Cotran & Kumar Pathologic Basis of Disease: "Necrosis is a pathologic process that is the consequence of severe injury. The main causes of necrosis include loss of oxygen supply (ischemia), exposure to microbial toxins, burns and other forms of chemical and physical injury, and unusual situations in which active proteases leak out of cells and damage surrounding tissues (as in pancreatitis)."
Causes of Necrosis
The principal triggers of necrosis include:
- Ischemia - Loss of blood supply deprives cells of oxygen and nutrients, causing failure of mitochondrial oxidative phosphorylation and ATP depletion. Without ATP, the sodium-potassium pump fails, leading to intracellular sodium and water accumulation, cell swelling, and eventually membrane rupture.
- Physical agents - Severe heat, cold, radiation, and trauma physically destroy cell membranes and denature proteins.
- Chemical and toxic agents - Heavy metals, cyanide, carbon tetrachloride, and bacterial toxins damage organelle membranes and disrupt cellular metabolism.
- Microbial infections - Certain bacteria and fungi release toxins that lyse membranes directly.
- Enzymatic self-digestion - In acute pancreatitis, activated digestive enzymes escape into surrounding pancreatic tissue and cause fat necrosis.
A key biochemical trigger is ATP depletion. As noted in Robbins Basic Pathology, reduced activity of plasma membrane ATP-dependent sodium pumps results in intracellular accumulation of sodium, osmotic gain of water, cell swelling, and dilation of the endoplasmic reticulum (ER). Compensatory anaerobic glycolysis leads to lactic acid accumulation and decreased intracellular pH, further disabling enzymatic processes. Ultimately, irreversible damage to mitochondrial and lysosomal membranes occurs, and the cell undergoes necrosis.
The "point of no return" - where reversible injury becomes irreversible - is characterized by two phenomena: (1) inability to reverse mitochondrial dysfunction even after the original injury is removed, and (2) profound disturbances in membrane function, particularly of the lysosomal membrane.
Morphology of Necrosis
Necrosis produces characteristic histological and ultrastructural changes that pathologists use to identify and classify tissue damage.
Cytoplasmic changes:
- Increased eosinophilia - Necrotic cells stain more intensely pink (eosinophilic) with H&E staining. This is due partly to loss of cytoplasmic RNA (which normally binds the blue hematoxylin dye) and partly to accumulation of denatured proteins that bind eosin.
- Glassy, homogeneous appearance - Loss of glycogen granules causes the cytoplasm to appear smooth and glassy compared to viable cells.
- Vacuolation ("moth-eaten" appearance) - When lysosomal enzymes digest cellular organelles, the cytoplasm becomes vacuolated.
- Myelin figures - Dead cells may be replaced by large whorled phospholipid precipitates called myelin figures, which are either phagocytosed or degraded into fatty acids. Calcium may bind these fatty acids, leading to dystrophic calcification.
Nuclear changes - The nucleus undergoes one of three patterns:
- Pyknosis - Nuclear shrinkage and increased basophilia; the DNA condenses into a dark, shrunken mass.
- Karyorrhexis - The pyknotic nucleus fragments into pieces.
- Karyolysis - The basophilia of the nucleus fades due to DNase-mediated digestion of DNA; within 1-2 days, the nucleus may completely dissolve.
Ultrastructural changes: By electron microscopy, necrotic cells show discontinuities in plasma and organelle membranes, marked dilation of mitochondria with large amorphous intramitochondrial densities, intracytoplasmic myelin figures, and aggregates of fluffy material representing denatured protein.
Inflammatory Response
A hallmark of necrosis is the inflammatory reaction it provokes in surrounding tissue. When cells undergo necrotic death, their membranes rupture and intracellular contents spill into the extracellular space. Among these contents are damage-associated molecular patterns (DAMPs), including ATP released from damaged mitochondria, uric acid (a breakdown product of DNA), and other molecules normally confined within healthy cells. These molecules are recognized by receptors on macrophages and other cells, triggering phagocytosis of debris and production of cytokines that amplify inflammation. Inflammatory cells (mainly neutrophils initially, then macrophages) produce additional proteolytic enzymes, and the combination of phagocytosis and enzymatic digestion eventually clears the necrotic debris.
This inflammatory response, while necessary for cleanup and repair, can itself cause collateral tissue damage - a key reason why necrosis is considered pathologically harmful.
Clinical Significance: Biomarkers
The leakage of intracellular proteins from necrotic cells is the basis for widely used blood tests. When myocardial cells undergo necrosis during a heart attack, they release cardiac-specific troponins (cTnI, cTnT) into the bloodstream. These can be detected as early as 2 hours after myocardial cell death - well before histological changes of infarction become visible. Similarly, hepatocyte necrosis releases transaminases (ALT, AST), and bile duct damage elevates alkaline phosphatase. These biomarkers are now central to clinical diagnosis of organ-specific injury.
Patterns of Tissue Necrosis
When large numbers of cells die in a tissue or organ, distinct morphological patterns of necrosis emerge, each reflecting the type of injury and tissue involved:
-
Coagulative necrosis - The most common type. The architecture of dead tissue is preserved for days because injury denatures both structural proteins and enzymes, blocking proteolysis. Ischemic infarcts (except in the brain) show coagulative necrosis. The tissue appears firm and eosinophilic with "ghost" outlines of cells.
-
Liquefactive necrosis - Characterized by enzymatic digestion of dead cells, transforming tissue into a viscous liquid. Seen in focal bacterial and fungal infections (pus is creamy yellow due to neutrophils) and, notably, in brain infarcts - because the CNS is rich in lipids and digestive enzymes but has little structural protein to maintain architecture.
-
Caseous necrosis - Named for its soft, cheese-like gross appearance, this pattern is most characteristic of tuberculosis. Microscopically, the area shows a structureless collection of fragmented cells and amorphous debris enclosed within epithelioid macrophages (granuloma).
-
Fat necrosis - Focal areas of fat destruction, typically seen in acute pancreatitis. Released pancreatic lipases digest fat cells, and the released fatty acids combine with calcium to form chalky-white deposits (saponification).
-
Gangrenous necrosis - A clinical term applied to a limb or organ with ischemic coagulative necrosis. "Wet gangrene" refers to superimposed bacterial infection adding liquefactive necrosis.
-
Fibrinoid necrosis - Seen in immune-mediated vascular diseases; immune complexes deposit in vessel walls along with leaked plasma proteins, creating a bright-pink ("fibrinoid") appearance on H&E staining.
Part II: Apoptosis
Definition and General Concept
Apoptosis is a fundamentally different form of cell death - one that is programmed, active, and energy-dependent. Rather than being an accident imposed by external injury, apoptosis is an internally activated program of cell self-destruction that the cell itself executes using its own enzymatic machinery. The term comes from Greek, meaning "falling off" (as leaves fall from a tree), coined by Kerr, Wyllie, and Currie in 1972 to describe the orderly fragmentation they observed in dying cells.
According to Robbins Basic Pathology: "Apoptosis is a pathway of cell death in which cells activate enzymes that degrade the cells' own nuclear DNA and nuclear and cytoplasmic proteins. Fragments of the apoptotic cells then break off, giving the appearance that is responsible for the name. The plasma membrane of the apoptotic cell remains intact, but the membrane is altered in such a way that the fragments, called apoptotic bodies, are recognized and rapidly phagocytosed by macrophages."
The critical distinction from necrosis: apoptotic cell fragments are cleared before cellular contents leak out, so apoptosis does not elicit an inflammatory reaction.
Physiological Roles of Apoptosis
Apoptosis is not only a response to pathological injury - it is a normal and necessary process throughout life:
- Embryonic development - Apoptosis sculpts organs and limbs. For example, the digits of the hand and foot are separated by the death of interdigital web cells.
- Homeostatic tissue turnover - In proliferative tissues such as intestinal epithelium and lymph nodes, cells undergo apoptosis to balance new cell production. Without this, tissues would overgrow.
- Hormone-dependent tissue involution - After estrogen withdrawal, the uterine endometrium involutes via apoptosis of glandular cells.
- Immune regulation - After an immune response, excess activated lymphocytes die by apoptosis to prevent chronic inflammation. Autoreactive T cells that could cause autoimmune disease are also eliminated by apoptosis in the thymus (negative selection).
- Cytotoxic T lymphocyte (CTL) killing - CTLs kill virus-infected and tumor cells by triggering apoptosis via perforin/granzyme or Fas-FasL mechanisms.
Pathological Causes of Apoptosis
Apoptosis also eliminates cells with dangerous or irreparable abnormalities:
- DNA damage - Radiation, chemotherapy, and oxidative stress cause DNA breaks. If damage cannot be repaired, p53 activation drives apoptosis (via BH3-only proteins), preventing the damaged cell from proliferating and potentially becoming cancerous.
- Protein misfolding (ER stress) - Accumulation of misfolded proteins in the ER triggers the unfolded protein response; if prolonged, this activates apoptosis.
- Viral infections - Certain viruses induce apoptosis of infected cells; cytotoxic T cells also kill infected cells by triggering caspase activation.
- Therapeutic drugs - Most cancer chemotherapeutic agents kill tumor cells by inducing apoptosis.
Morphology of Apoptosis
The morphological features of apoptosis are distinct from necrosis and can be recognized by light and electron microscopy:
- Cell shrinkage - The cell decreases in size; the cytoplasm becomes dense and eosinophilic, and organelles are more tightly packed. This is the opposite of necrosis, where early cell swelling (oncosis) is the first change.
- Chromatin condensation - The most characteristic feature. Chromatin aggregates peripherally under the nuclear membrane into dense crescentic masses. The nucleus may fragment into two or more pieces.
- Membrane blebbing and apoptotic body formation - The cell surface shows extensive blebbing. The cell then fragments into membrane-bound apoptotic bodies composed of cytoplasm and tightly packed organelles, with or without nuclear fragments.
- Phagocytosis - Apoptotic bodies expose phosphatidylserine ("eat-me" signals) on the outer leaflet of their membrane, which is recognized by phagocyte receptors. Macrophages rapidly engulf and digest the bodies via lysosomal enzymes.
- No inflammation - Because the plasma membrane remains intact until phagocytosis and cellular contents never spill into the extracellular space, no DAMPs are released and no inflammatory reaction is triggered.
On H&E staining, apoptotic cells appear as round or oval masses of intensely eosinophilic cytoplasm with fragments of dense nuclear chromatin. Because the process is rapid and fragments are quickly phagocytosed, apoptosis can be easily missed in tissue sections.
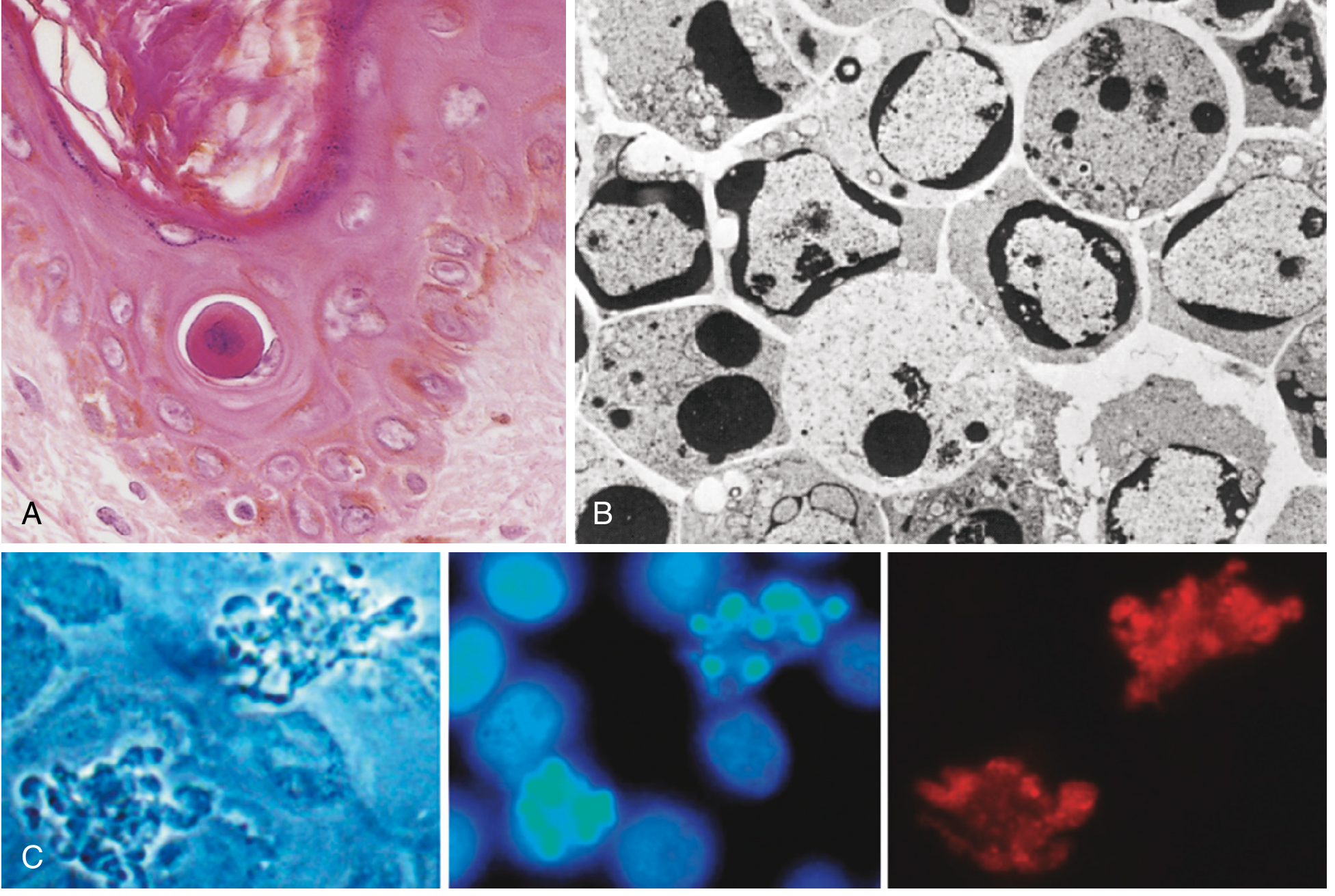
Morphologic features of apoptosis. (A) H&E stain showing reduced, eosinophilic apoptotic cell with condensed nucleus. (B) Electron micrograph showing peripheral chromatin condensation crescents. (C) Phase contrast and fluorescence images showing blebbing, nuclear DNA fragmentation, and caspase-3 activation. - Robbins, Cotran & Kumar Pathologic Basis of Disease
Biochemical Mechanisms of Apoptosis: The Caspase Cascade
The central executioners of apoptosis are caspases - a family of cysteine proteases that cleave proteins at aspartate residues. They exist as inactive precursors (procaspases) and are activated in a cascade. Caspases are divided into:
- Initiator caspases (caspase-8, caspase-9) - Activated first, depending on the pathway.
- Executioner caspases (caspase-3, caspase-6, caspase-7) - Activated by initiator caspases; these cleave cytoskeletal proteins, nuclear lamins, and activate DNases that fragment DNA.
There are two main apoptotic pathways:
1. The Mitochondrial (Intrinsic) Pathway
This is the most common pathway, triggered by intracellular stress signals (DNA damage, growth factor withdrawal, protein misfolding, oxidative stress). Its regulation centers on the BCL-2 family of proteins, which includes:
- Anti-apoptotic proteins: BCL-2, BCL-XL, MCL-1 (located in the outer mitochondrial membrane; they block cytochrome c release)
- Pro-apoptotic effectors: BAX, BAK (form channels in the mitochondrial membrane when activated)
- BH3-only sensors: BID, BAD, PUMA, NOXA (detect cellular stress and tip the balance toward apoptosis by inhibiting BCL-2 or directly activating BAX/BAK)
When stress signals overwhelm survival signals, BH3-only proteins activate BAX/BAK, which form pores in the outer mitochondrial membrane, releasing cytochrome c and other pro-apoptotic proteins (e.g., SMAC/DIABLO, AIF) into the cytoplasm. Cytochrome c binds APAF-1 (apoptotic protease activating factor-1) and, together with cytochrome c and dATP, forms a multimeric complex called the apoptosome, which activates caspase-9 (initiator). Caspase-9 then activates executioner caspases-3 and -6, leading to cell demolition.
2. The Death Receptor (Extrinsic) Pathway
This pathway is triggered by external death signals binding to cell-surface death receptors belonging to the TNF receptor superfamily. The most important examples are:
- Fas (CD95/APO-1) - binds FasL (Fas ligand, expressed on cytotoxic T lymphocytes and some other cells)
- TNFR1 - binds tumor necrosis factor (TNF)
When FasL binds and cross-links Fas receptors, the receptors cluster and recruit an adaptor protein called FADD (Fas-associated death domain protein) via death domain interactions. FADD recruits procaspase-8, and the resulting complex (the DISC - death-inducing signaling complex) enables autocatalytic activation of caspase-8. Active caspase-8 then directly cleaves and activates executioner caspases-3, -6, and -7.
A key regulatory protein in this pathway is FLIP (FLICE-inhibitory protein), which competes with procaspase-8 for FADD binding and inhibits this pathway. Many viruses encode FLIP-like proteins to protect infected cells from Fas-mediated killing.
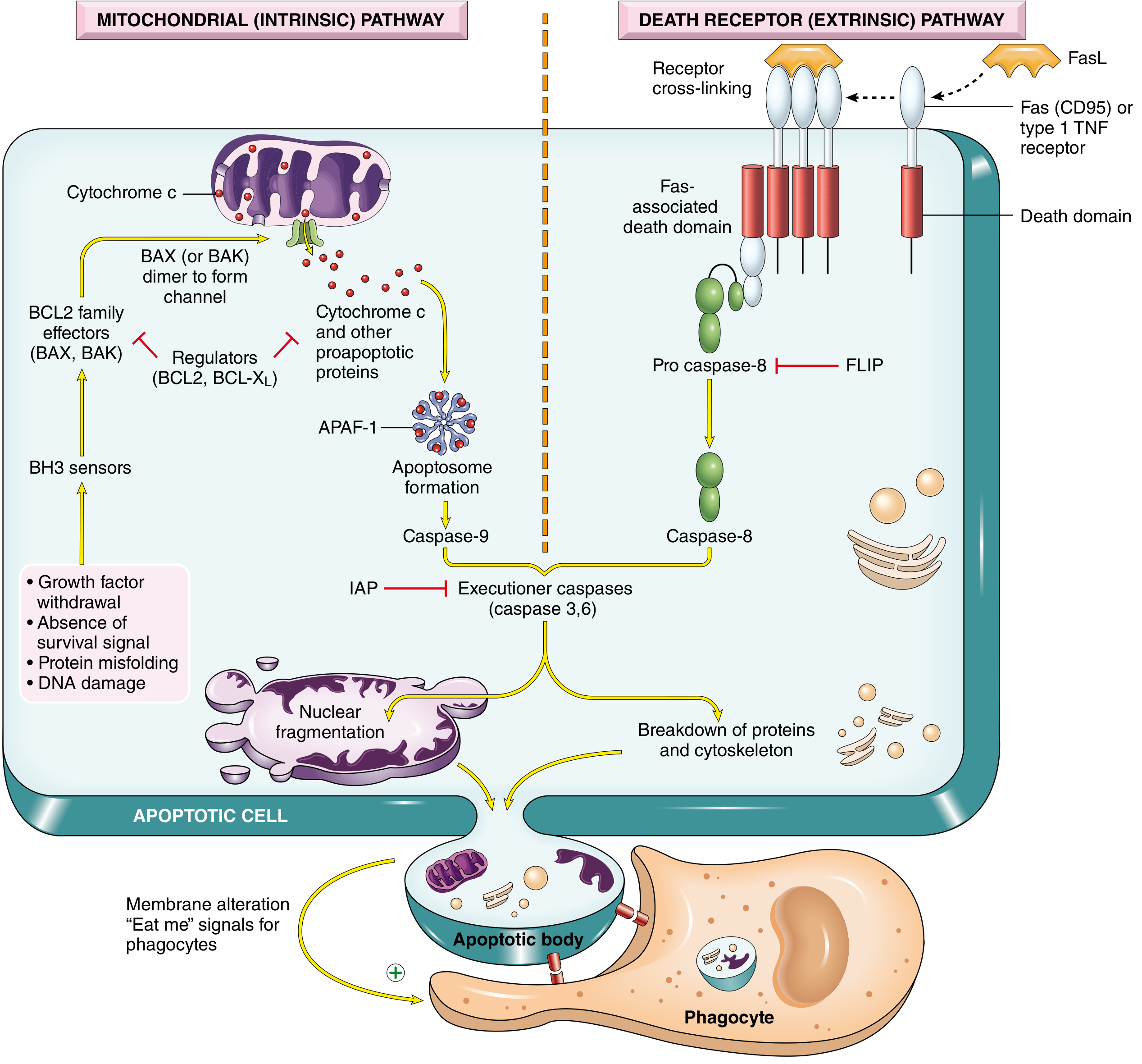
The two main apoptosis pathways. Left (mitochondrial/intrinsic): Growth factor withdrawal, DNA damage, and protein misfolding activate BH3 sensors → BAX/BAK channel formation → cytochrome c release → apoptosome → caspase-9 → executioner caspases. Right (death receptor/extrinsic): FasL or TNF binds death receptors → FADD recruitment → caspase-8 activation → executioner caspases. Both converge on nuclear fragmentation and apoptotic body formation. - Robbins, Cotran & Kumar Pathologic Basis of Disease
DNA Fragmentation
The executioner caspases activate a caspase-activated DNase (CAD) that cleaves DNA between nucleosomes, generating fragments that are multiples of ~180 base pairs. On gel electrophoresis, this produces the characteristic "DNA ladder" pattern - a biochemical hallmark of apoptosis used in laboratory detection.
Inhibitors of Apoptosis
Given the dangers of uncontrolled apoptosis, the cell has built-in brakes:
- IAPs (Inhibitors of Apoptosis Proteins) such as XIAP directly bind and inhibit executioner caspases.
- BCL-2 and BCL-XL at the mitochondrial membrane prevent cytochrome c release.
- Survival signals (growth factors, cytokines) act through kinase pathways (e.g., PI3K/AKT) that phosphorylate and inactivate pro-apoptotic proteins.
- Cancer cells frequently exploit these mechanisms to escape apoptosis - BCL-2 overexpression is a driver of follicular lymphoma, for example.
Clinical Significance of Apoptosis
Insufficient apoptosis (too little cell death):
- Cancer - Mutations in TP53 (p53), overexpression of BCL-2, or loss of death receptors allow damaged cells to survive and accumulate mutations.
- Autoimmune disease - Failure to eliminate autoreactive lymphocytes.
- Viral persistence - Viruses that block apoptosis allow infected cells to survive and replicate indefinitely.
Excessive apoptosis (too much cell death):
- Neurodegenerative diseases - In Alzheimer's, Parkinson's, and amyotrophic lateral sclerosis (ALS), neurons undergo apoptotic death.
- AIDS - HIV-infected T cells and even uninfected bystander CD4+ cells undergo apoptosis, depleting the immune system.
- Ischemia-reperfusion injury - Reperfusion of ischemic tissue paradoxically triggers apoptosis through ROS and cytochrome c release.
- Graft-versus-host disease - Donor cytotoxic T cells kill host cells by Fas-FasL-mediated apoptosis.
Part III: Comparing Apoptosis and Necrosis
Having examined each form of cell death in detail, a systematic comparison highlights the key distinctions:
| Feature | Apoptosis | Necrosis |
|---|---|---|
| Type of death | Programmed, physiologic or pathologic | Always pathologic (accidental) |
| Trigger | Physiological signals, DNA damage, growth factor withdrawal, death receptor activation | Severe ischemia, toxins, physical injury, infection |
| Energy requirement | Active, requires ATP | Passive, occurs when ATP is depleted |
| Cell size | Shrinkage (cell becomes smaller) | Swelling (oncosis - cell enlarges and bursts) |
| Plasma membrane | Intact until phagocytosis | Ruptured early |
| Nucleus | Chromatin condensation (pyknosis), fragmentation | Pyknosis → karyorrhexis → karyolysis |
| Cytoplasm | Dense, eosinophilic, organelles intact | Vacuolated, "moth-eaten," organelles disrupted |
| Cell fragments | Membrane-bound apoptotic bodies | Cellular debris (no membrane wrapping) |
| DNA degradation | Internucleosomal ("ladder" pattern on gel) | Random, non-specific degradation |
| Caspases | Central executioners (caspase-3, -8, -9) | Not primarily involved |
| Inflammatory response | None (contents not leaked, no DAMPs released) | Prominent (DAMPs trigger inflammation) |
| Fate of dead cell | Phagocytosed rapidly without trace | Persists until cleared by leukocytes |
| Scope | Single cells or small clusters | Often affects large areas of tissue |
| Clinical examples | Thymic selection, hormone-dependent involution, cancer chemotherapy effect | Myocardial infarction, gangrene, tuberculosis (caseous), brain infarct |
| BCL-2 family involvement | Yes - central regulatory role | Not a primary mechanism |
| Cytochrome c release | Yes (mitochondrial pathway) | As late, nonspecific event only |
Key Conceptual Distinctions
Control vs. chaos: Apoptosis is a tightly regulated, internally directed program. The cell orchestrates its own demolition in a way that protects neighbors. Necrosis is uncontrolled dissolution - the cell is overwhelmed and falls apart passively.
Membrane integrity: This is perhaps the single most important structural distinction. In apoptosis, the plasma membrane stays intact throughout most of the process, confining cellular contents until phagocytosis. In necrosis, membrane disruption is an early event, releasing the cell's contents into the surrounding tissue.
Inflammation: Because of the membrane integrity difference, apoptosis is "immunologically silent" while necrosis is inherently inflammatory. This has enormous biological implications - organisms use apoptosis precisely where inflammation would be dangerous (e.g., eliminating self-reactive lymphocytes, pruning neurons during brain development).
Energy dependence: Apoptosis requires ATP to fuel the caspase cascade, DNA fragmentation, membrane blebbing, and phosphatidylserine externalization ("eat-me" signals). Necrosis, by contrast, is often a consequence of ATP depletion - the cell cannot maintain membrane pumps and structural integrity.
Scale and reversibility: Necrosis usually kills large areas of tissue; apoptosis eliminates individual cells. An important nuance is that these pathways are not always mutually exclusive. In some circumstances (e.g., ischemia, oxidative stress, radiation), both pathways can be active simultaneously. Apoptosis can progress to "secondary necrosis" if the dead cell is not promptly phagocytosed. The concept of necroptosis - a programmed form of necrosis triggered through death receptors - further blurs the boundary, sharing features of both.
Part IV: Overlap and Modern Perspectives
Modern cell biology has revealed that the boundary between apoptosis and necrosis is not always sharp. As Robbins, Cotran & Kumar note: "In some situations, cell death may show morphologic features of both apoptosis and necrosis, and apoptosis may progress to necrosis, so the distinctions may not be absolute."
Necroptosis is the clearest example of this overlap: it is triggered by the same death receptors (Fas, TNFR1) that activate apoptosis, but when caspase-8 is inhibited, a kinase cascade involving RIPK1 and RIPK3 activates the pore-forming protein MLKL, causing cell lysis similar to necrosis. This pathway is important in some infections (where viruses inhibit caspases) and in inflammatory bowel disease.
Pyroptosis is another programmed necrotic pathway, triggered by inflammasome activation (e.g., in response to bacterial toxins) and resulting in rapid lysis and massive IL-1β/IL-18 release.
These discoveries have shifted the paradigm: cell death is not simply a binary choice between orderly apoptosis and messy necrosis. It is a spectrum of regulated and unregulated pathways, each with distinct molecular machinery and functional consequences.
Conclusion
Apoptosis and necrosis represent two fundamentally different strategies by which cells die, each with profound implications for health and disease. Necrosis is a catastrophic, inflammation-provoking dissolution of cells overwhelmed by severe injury - characterized by membrane rupture, organelle swelling, enzymatic digestion, and tissue inflammation. Apoptosis is a precise, energy-dependent program of controlled self-destruction - characterized by cell shrinkage, chromatin condensation, membrane-intact fragmentation, and silent phagocytic clearance.
The caspase cascade, BCL-2 family proteins, cytochrome c release, and death receptors define the molecular architecture of apoptosis. The failure of ATP production, lysosomal leakage, and membrane breakdown define the path of necrosis. Both processes can contribute to diseases ranging from myocardial infarction and stroke to cancer, neurodegeneration, and autoimmunity.
An understanding of these two pathways - their triggers, mechanisms, morphological features, and clinical biomarkers - is foundational to modern pathology, pharmacology, and therapeutic development. Drugs that target the apoptotic machinery (BCL-2 inhibitors like venetoclax for leukemia, for example) are already in clinical use, and many more are in development. The cell's decision between life and death, and between orderly and chaotic death, sits at the heart of human disease.
Sources:
- Robbins & Kumar Basic Pathology (9780323790185), Chapter 1 - pp. 19-31, 37-41
- Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528), Chapter 2 - pp. 50-62
- Kerr JFR, Harmon BV. Definition and incidence of apoptosis: a historical perspective. In Tomei LD, Cope FO, eds. Apoptosis: The Molecular Basis of Cell Death. Cold Spring Harbor Laboratory Press; 1991.
This is a shared conversation. Sign in to Orris to start your own chat.