60 years old hospitalised for chest pain. 2months ago. He has blockage in left artery. PCI with placement of a was discharged with medical term. It may include hypertension type 2 diabetes and generative joint disease. Patient has been taking all the medication has not chest pain. Diffuse muscle aches and cramps especially after exercise. Elevated serum kinase
Checking Date & Time
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Searching PubMed
statin myopathy management discontinuation rechallenge
I now have all the information needed to deliver a comprehensive, well-structured clinical answer.
Clinical Analysis: Post-PCI Patient with Muscle Symptoms
The Diagnosis: Statin-Associated Myopathy (SAM)
This is the classic presentation. The patient had a PCI 2 months ago for LAD occlusion and was discharged on a high-intensity statin (almost certainly atorvastatin 40–80 mg or rosuvastatin 20–40 mg — standard of care post-ACS/PCI). He now presents with:
| Finding | Significance |
|---|---|
| Diffuse muscle aches and cramps | Myalgia / myopathy |
| Worse after exercise | Hallmark of statin myopathy |
| Elevated serum creatine kinase (CK) | Confirms myopathy (not just myalgia) |
| No chest pain | Cardiac cause excluded |
Spectrum of Statin Muscle Toxicity
Statin muscle effects exist on a continuum:
| Term | Definition | CK Level |
|---|---|---|
| Myalgia | Muscle pain/weakness, no CK elevation | Normal |
| Myopathy | Muscle symptoms + elevated CK | >10× ULN |
| Rhabdomyolysis | Severe: CK >10× ULN + elevated creatinine + myoglobinuria | >10× ULN + organ involvement |
This patient has elevated CK → myopathy, not just myalgia.
— Harrison's Principles of Internal Medicine 22E, p. 60–61
Mechanism
Statins inhibit HMG-CoA reductase → reduce hepatic cholesterol synthesis → but also deplete mevalonate pathway intermediates including:
- Ubiquinone (Coenzyme Q10) — essential for mitochondrial electron transport in skeletal muscle
- Heme A — part of cytochrome c oxidase
This impairs mitochondrial ATP production in skeletal muscle, explaining the exercise-induced worsening.
— Biochemistry, 8th ed Lippincott Illustrated Reviews
Risk Factors for Statin-Induced Myopathy
This patient has multiple compounding risk factors:
- Age 60 (risk increases with age, especially >80)
- Type 2 Diabetes — associated with altered lipid metabolism and drug clearance
- Post-PCI → likely on high-intensity statin (higher dose = higher plasma concentration = higher risk)
- Degenerative joint disease → may be on NSAIDs, which can interact
- Hypertension → may be on amlodipine (CYP3A4 inhibitor — increases simvastatin/lovastatin plasma levels)
Other general risk factors include: renal/hepatic dysfunction, hypothyroidism, fibrate co-administration (especially gemfibrozil), macrolide antibiotics, azole antifungals, cyclosporine, amiodarone.
— Goodman & Gilman's, p. 3035–3037
Drug Interactions to Check Immediately
| Drug | Statin interaction |
|---|---|
| Gemfibrozil (fibrate) | ↑ statin plasma levels by ~38% — most dangerous |
| Amlodipine (CCB for HTN) | Mild CYP3A4 inhibition — raises simvastatin |
| Amiodarone | ↑ statin levels |
| Cyclosporine | Major CYP3A4 inhibitor |
Management Approach (Evidence-Based)
Step 1 — Assess severity
-
Mild-moderate symptoms (this patient's likely category if CK <10× ULN):
- Stop the statin
- Wait until symptoms resolve (~2 months)
- Rechallenge with lower dose of same statin OR switch to another statin
-
Severe/rhabdomyolysis (CK >10× ULN + elevated creatinine or myoglobinuria):
- Stop statin immediately
- Check CK, creatinine/GFR, urine myoglobin
- Admit and IV hydrate to prevent acute kidney injury
- Refer to lipidologist before restarting — Goldman-Cecil Medicine, Table 190-5
Step 2 — Critical: Do NOT permanently abandon statin therapy
This patient had a recent MI/ACS + PCI — statins are Class I indication for secondary prevention. The cardiovascular benefit far outweighs myopathy risk.
Options after myopathy resolves:
| Strategy | Details |
|---|---|
| Lower dose | Switch to moderate-intensity statin (atorvastatin 20 mg, rosuvastatin 10 mg) |
| Less frequent dosing | Rosuvastatin or atorvastatin can be given every other day or 2×/week (both are long-acting) |
| Switch statin | Pravastatin/fluvastatin not metabolized by CYP3A4 → less interaction risk |
| Add ezetimibe | Additive LDL-C lowering of ~18% → allows lower statin dose |
| PCSK9 inhibitor | If statin truly intolerable — evolocumab/alirocumab are highly effective alternatives |
📌 Key evidence: Placebo-controlled trials show ~75–80% of patients reporting statin intolerance can tolerate atorvastatin 20 mg on blinded rechallenge. Two-thirds of patients successfully restart statin therapy. — Goldman-Cecil Medicine, p. 13614
A 2025 systematic review and meta-analysis (PMID: 41379822) evaluated safety of statin vs. non-statin lipid-lowering options in patients with prior statin-associated muscle symptoms — relevant for guiding therapy if rechallenge fails.
Coenzyme Q10 — Should it be Added?
Statins reduce circulating CoQ10 levels. Some propose supplementation to reduce myopathy:
- A meta-analysis evaluating CoQ10 on statin-induced myopathy found some reduction in muscle pain and CK with CoQ10 (30 mg, 30 days of supplementation)
- Evidence is moderate quality; not universally recommended but reasonable to try
- CoQ10 may also benefit this patient's post-MI cardiac function — Katzung's Basic and Clinical Pharmacology, 16th ed, p. 435
Summary for This Patient
Diagnosis: Statin-associated myopathy (confirmed by elevated CK)
Cause: High-intensity statin post-PCI; possibly potentiated by
age, T2DM, or drug interactions (check amlodipine, any fibrate)
Immediate Rx: 1. Discontinue statin
2. Measure CK, creatinine, urine myoglobin
3. If CK >10× ULN + renal involvement → admit + IV fluids
4. Investigate other causes (hypothyroidism, polymyositis)
Long-term Rx: Restart statin at lower dose or different statin after
resolution (mandatory — secondary prevention post-MI)
Consider: every-other-day rosuvastatin + ezetimibe
Consider: CoQ10 supplementation
If truly statin-intolerant → PCSK9 inhibitor
⚠️ Key teaching point: A patient 2 months post-PCI on high-intensity statin presenting with exercise-induced diffuse myalgias + elevated CK = statin myopathy until proven otherwise. Always check CK, creatinine, and urine myoglobin to rule out rhabdomyolysis.
Why stop statin
Checking Date & Time
Great question — and it's a crucial one because stopping the statin feels counterintuitive in a post-PCI patient who needs it.
Why Stop the Statin in Statin-Associated Myopathy?
1. To Confirm the Diagnosis
The only way to prove the statin is the cause of the muscle symptoms is to stop it and see if symptoms resolve. Muscle aches have many causes (hypothyroidism, polymyositis, degenerative joint disease, deconditioning). If symptoms don't resolve within 2 months of stopping, the statin is NOT the cause — look elsewhere.
2. To Prevent Progression to Rhabdomyolysis
This is the critical safety reason. The spectrum is:
Myalgia → Myopathy → Rhabdomyolysis → Acute Kidney Injury → Death
If you continue the statin while CK is already elevated:
- Ongoing muscle fiber destruction continues
- CK can rise to >10× ULN
- Myoglobin released from damaged muscle precipitates in renal tubules → acute tubular necrosis
- Can cause acute renal failure and potentially death
The statin is literally the ongoing toxic insult — continuing it is like leaving your hand on a hot stove while treating the burn.
3. The Statin Itself Cannot Reduce CK or Repair Muscle While It's Still Causing the Damage
The mechanism (CoQ10/heme A depletion → mitochondrial dysfunction in skeletal muscle) is ongoing as long as the drug is present. Stopping allows mitochondrial function to recover and muscle fibers to heal.
4. Stopping is Temporary, Not Permanent
This is the most important nuance. Stopping ≠ stopping forever.
The statin is stopped temporarily to:
- Allow symptoms to resolve
- Rule out rhabdomyolysis
- Identify and correct any risk factors (drug interactions, hypothyroidism, dose)
- Rechallenge with a lower dose or different statin
For this post-PCI patient, the statin must be restarted — it reduces risk of another MI, stent thrombosis, and death. The goal is to find a statin strategy he can tolerate, not to abandon therapy.
In Short
| Reason | Consequence of NOT stopping |
|---|---|
| Confirm causality | Can't diagnose statin myopathy without a drug-free period |
| Prevent rhabdomyolysis | Ongoing CK rise → renal failure |
| Allow muscle recovery | Mitochondrial damage continues while statin is on board |
| Enable safe rechallenge | Need a baseline before restarting |
The statin is paused to protect the muscle, then reintroduced strategically — because the cardiovascular risk of being off a statin long-term in a post-MI patient far outweighs the temporary gap.
Why kinase is elevated
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Now I have the full biochemical chain. Here's the complete explanation:
Why Is Creatine Kinase (CK) Elevated?
The answer requires understanding two things: what CK is, and how statins damage muscle cells.
What Is Creatine Kinase?
CK is an intracellular enzyme found predominantly inside muscle cells (skeletal muscle, cardiac muscle, brain). Under normal conditions, the cell membrane is intact — CK stays inside the cell.
When muscle fibers are damaged or destroyed (necrosis), the cell membrane ruptures → intracellular contents, including CK, leak out into the bloodstream → serum CK rises.
Elevated serum CK = evidence of muscle cell membrane breakdown = myocyte death/injury
This is the same principle as elevated troponin in a heart attack (cardiomyocyte necrosis leaks troponin into blood).
How Do Statins Cause Muscle Cell Damage?
The mechanism starts in the mevalonate (cholesterol biosynthesis) pathway:
Acetyl-CoA
↓
HMG-CoA
↓ ← STATINS BLOCK HERE (inhibit HMG-CoA reductase)
Mevalonate
↓
Farnesyl pyrophosphate
↓ ↓
Cholesterol Ubiquinone (CoQ10)
Heme A
Statins are structural analogues of HMG-CoA and competitively inhibit HMG-CoA reductase. This doesn't just reduce cholesterol — it also depletes downstream products of the mevalonate pathway, specifically:
| Depleted product | Role in muscle |
|---|---|
| Ubiquinone (CoQ10) | Essential electron carrier in mitochondrial oxidative phosphorylation (Complex I→II→III) |
| Heme A | Component of cytochrome c oxidase (Complex IV) — final step of electron transport chain |
Both are critical for ATP production in skeletal muscle via oxidative phosphorylation.
The Chain of Events
Statin inhibits HMG-CoA reductase
↓
↓ Ubiquinone + ↓ Heme A in skeletal muscle mitochondria
↓
Impaired oxidative phosphorylation → ↓ ATP production in muscle cells
↓
Energy-starved muscle cells → mitochondrial dysfunction → cell membrane instability
↓
Muscle fiber necrosis (especially under metabolic stress — e.g., exercise)
↓
CK leaks from ruptured myocytes into bloodstream
↓
↑ Serum CK
This is why symptoms are worse after exercise — exercise demands more ATP, and muscles already depleted of CoQ10/Heme A cannot meet the demand, accelerating cell damage.
— Biochemistry, 8th ed Lippincott Illustrated Reviews, p. 628
Genetic Factor Worth Knowing
A polymorphism in the OATP1B1 transporter gene (SLCO1B1, variant 521 T>C) impairs hepatic uptake of statins — so more statin accumulates in the bloodstream and reaches skeletal muscle. This is used as a biomarker for simvastatin-induced myopathy, explaining why some patients are more susceptible than others at the same dose.
— Biochemistry, 8th ed Lippincott Illustrated Reviews, p. 628
Summary in One Sentence
Statins deplete CoQ10 and Heme A in skeletal muscle mitochondria → impaired ATP synthesis → muscle fiber necrosis → CK (an intracellular enzyme) leaks out of ruptured cells into the bloodstream → elevated serum CK.
Increased hepatocyte ldl
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
I have the key content. Here's the full explanation:
Why Do Statins Increase Hepatocyte LDL Receptors? (And How This Lowers Plasma LDL)
This is the primary therapeutic mechanism of statins — the muscle side effect is just an unfortunate consequence of the same pathway.
The Core Logic: The Liver Senses a Cholesterol Shortage
Statin blocks HMG-CoA reductase
↓
↓ Cholesterol synthesis inside hepatocytes (liver cells)
↓
Hepatocyte senses: "I am low on cholesterol — I need more"
↓
Compensatory (counterregulatory) response:
↑ Expression of LDL receptors on hepatocyte surface
↓
More LDL receptors grab LDL particles from the bloodstream
↓
↓ Plasma LDL-C (30–55% reduction)
— Harrison's Principles of Internal Medicine 22E, p. 3294
The Molecular Mechanism (SREBP Pathway)
When intracellular cholesterol falls inside the hepatocyte:
- SREBP (Sterol Regulatory Element-Binding Protein) — a transcription factor kept inactive when cholesterol is abundant — is released and activated
- SREBP travels to the nucleus and binds to the Sterol Regulatory Element (SRE) on DNA
- This upregulates transcription of the LDL receptor gene
- More LDL receptors are synthesized and inserted into the hepatocyte surface membrane
- These receptors bind circulating LDL particles via ApoB-100
- The LDL-receptor complex is endocytosed into the hepatocyte
- LDL is degraded in lysosomes → cholesterol recycled intracellularly
Visual Summary
LOW intracellular cholesterol in hepatocyte
↓
SREBP activated → nucleus → transcribes LDL receptor gene
↓
↑ LDL receptors on hepatocyte surface
↓
LDL from blood binds → endocytosed → degraded
↓
↓ Serum LDL-C
Important Nuances
| Point | Explanation |
|---|---|
| Dose-response is limited | Doubling the statin dose only gives ~6% additional LDL reduction (the receptor upregulation plateaus) |
| Ezetimibe works the same way | Blocks intestinal cholesterol absorption → less cholesterol delivered to liver → same SREBP activation → same LDL receptor upregulation (~18% further LDL reduction) — additive with statins |
| Familial Hypercholesterolaemia (FH) | Statins don't work well in homozygous FH because the LDL receptor gene is mutated — you can upregulate expression of a broken receptor all you like, it still won't clear LDL |
| PCSK9 | Normally degrades LDL receptors after endocytosis. PCSK9 inhibitors block this → receptors are recycled back to surface → even more LDL cleared |
Tying It Back to This Patient
This patient is post-PCI on a high-intensity statin specifically to maximally upregulate hepatic LDL receptors and drive plasma LDL-C below 70 mg/dL (secondary prevention target). The same drug that is therapeutically increasing his hepatic LDL receptors is simultaneously depleting CoQ10 in his skeletal muscle — causing his myopathy. Two consequences of blocking the same enzyme.
Renal calcium re absorption
Checking Date & Time
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Excellent diagrams. Here is the complete explanation:
Renal Calcium Reabsorption
Of the calcium filtered at the glomerulus, ~99% is reabsorbed along the nephron. Only ~1% is excreted in urine.
Where Along the Nephron?
| Segment | % Reabsorbed | Mechanism | Hormonal control |
|---|---|---|---|
| Proximal convoluted tubule (PCT) | ~65–75% | Passive, paracellular via claudin-2 channels | PTH-independent; follows Na⁺/H₂O |
| Thick ascending limb of Loop of Henle (TAL) | ~15–20% | Passive, paracellular via claudin-16 channels | Ca²⁺ sensing receptor (CaSR); PTH, calcitriol |
| Distal convoluted tubule (DCT) | ~10–15% | Active, transcellular | PTH, Vitamin D (calcitriol) — primary regulated site |
| Collecting duct | <5% | Minor | — |
— Campbell-Walsh Urology; Guyton & Hall Medical Physiology
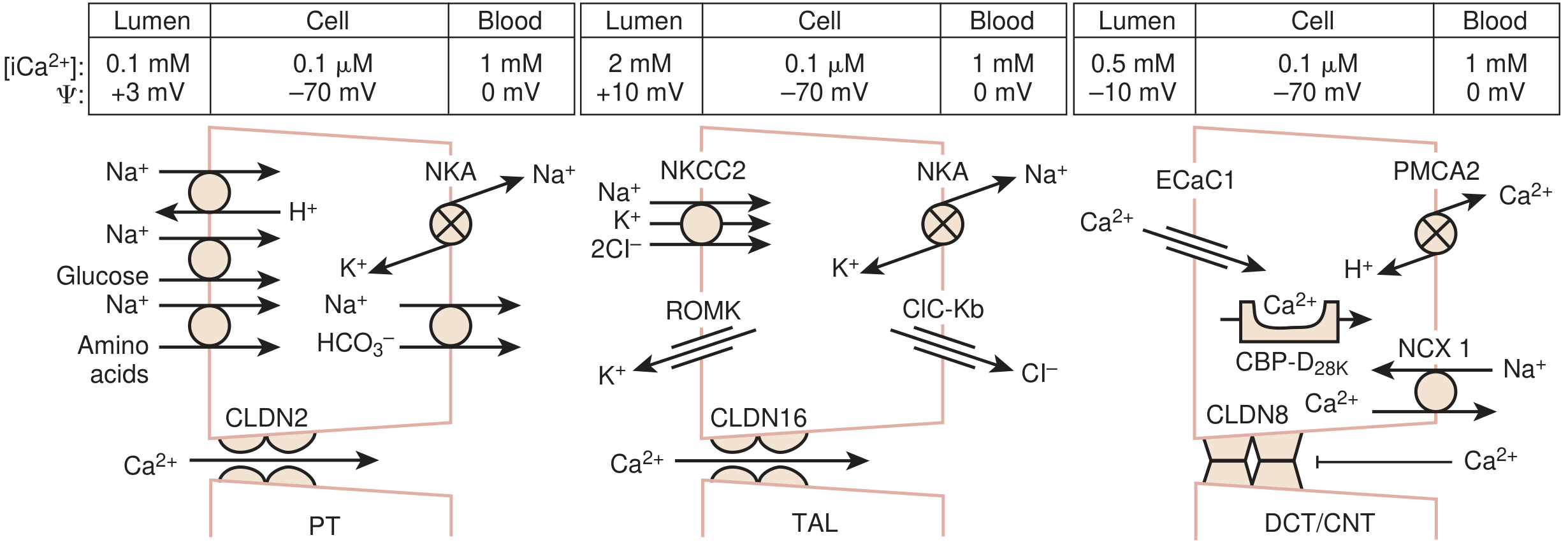
Diagrams
Figure 1 — Mechanisms at each segment (Guyton & Hall):
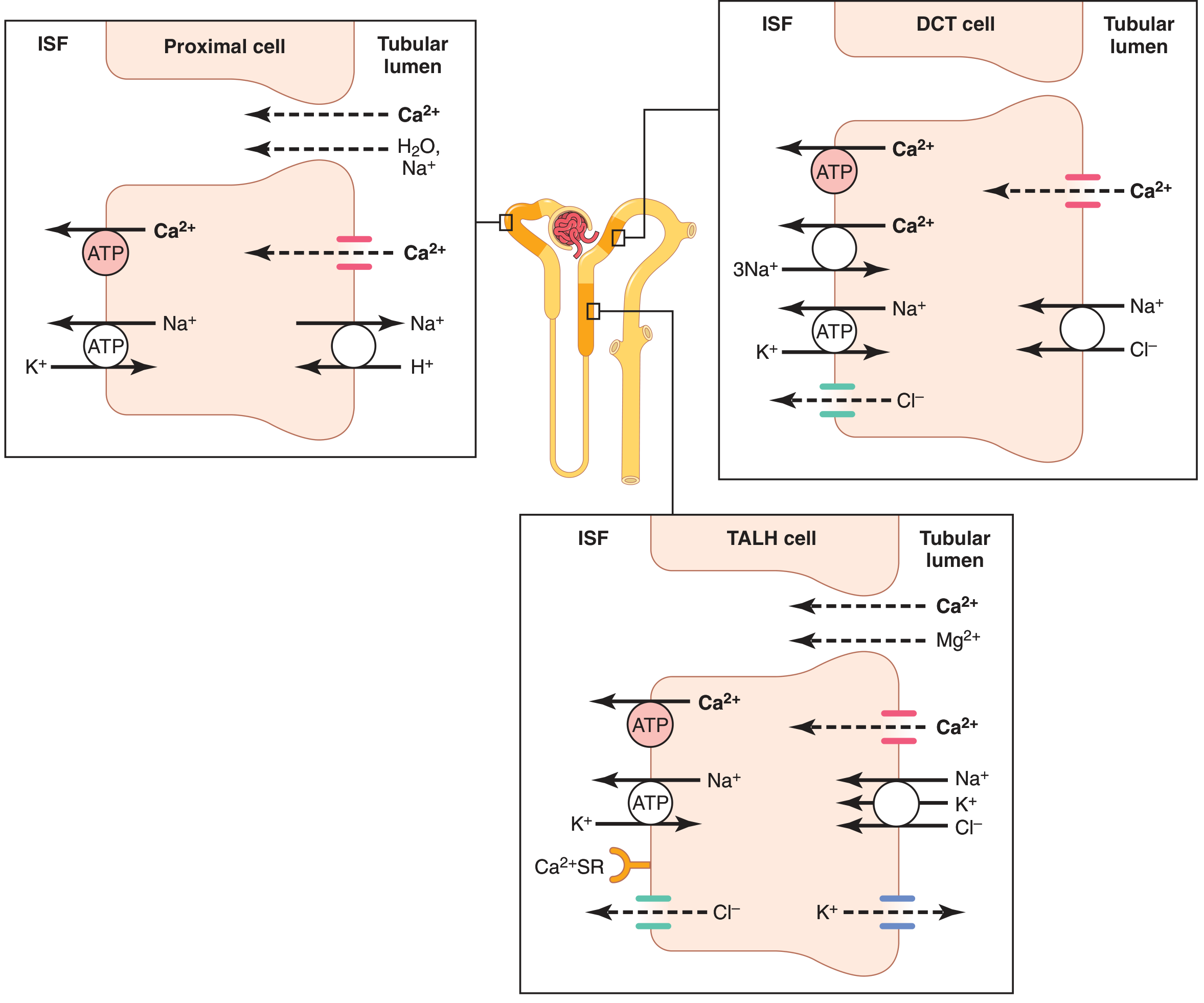
Figure 2 — Transporters at PT, TAL, and DCT/CNT (Campbell-Walsh):
Segment-by-Segment Mechanisms
1. Proximal Convoluted Tubule (PCT) — 65–75%
- Reabsorption is paracellular (between cells), driven by the lumen-positive voltage and water drag (solvent drag with Na⁺ and H₂O)
- Channel: Claudin-2 tight junction protein
- PTH-independent — calcium here simply follows sodium and water
- If volume is expanded (e.g., IV fluids, hypertension) → ↓ Na⁺/H₂O reabsorption → ↓ Ca²⁺ reabsorption → more calcium excreted
2. Thick Ascending Limb of Loop of Henle (TAL) — 15–20%
- Also paracellular via claudin-16 channels
- Driven by the lumen-positive voltage created by NKCC2 (Na⁺-K⁺-2Cl⁻ cotransporter) activity
- CaSR (Calcium Sensing Receptor) on basolateral membrane monitors blood calcium:
- High blood Ca²⁺ → activates CaSR → inhibits NKCC2 → reduces lumen-positive voltage → ↓ Ca²⁺ reabsorption → more Ca²⁺ excreted (direct feedback)
- PTH and calcitriol increase reabsorption here
💡 Loop diuretics (furosemide) block NKCC2 → abolish the lumen-positive voltage → block Ca²⁺ reabsorption at TAL → hypercalciuria. Used to treat hypercalcaemia.
3. Distal Convoluted Tubule (DCT) — 10–15%
- Transcellular (active) — the fine-tuning site
- Step 1 (Lumen → Cell): Ca²⁺ enters via ECaC1/TRPV5 channel on luminal membrane
- Step 2 (Inside cell): Bound by calbindin-D28K (keeps intracellular Ca²⁺ low, allowing continued inward movement)
- Step 3 (Cell → Blood): Exits basolaterally via:
- PMCA (Ca²⁺-ATPase pump) — active, against concentration gradient
- NCX1 (Na⁺/Ca²⁺ exchanger) — secondary active
💡 Thiazide diuretics block NaCl cotransporter (NCC) in DCT → intracellular Na⁺ falls → NCX1 works harder (more Na⁺ in, more Ca²⁺ out) → ↓ intracellular Ca²⁺ → ↑ ECaC1 entry from lumen → ↑ Ca²⁺ reabsorption → hypocalciuria. Used to prevent calcium kidney stones.
Hormonal Regulation Summary
| Hormone | Site of Action | Effect |
|---|---|---|
| PTH ↑ | TAL + DCT | ↑ Ca²⁺ reabsorption, ↓ Ca²⁺ excretion |
| PTH ↑ | PCT | Stimulates 1α-hydroxylase → activates Vitamin D |
| Calcitriol (Vit D) | DCT | ↑ calbindin-D28K → ↑ transcellular Ca²⁺ reabsorption |
| Calcitonin | TAL + DCT | ↑ reabsorption (minor role vs. PTH) |
| CaSR activation (↑ blood Ca²⁺) | TAL | Inhibits reabsorption → ↑ excretion |
| Metabolic alkalosis | DCT | ↑ reabsorption → ↓ Ca²⁺ excretion |
| Metabolic acidosis | DCT | ↓ reabsorption → ↑ Ca²⁺ excretion |
— Guyton & Hall Textbook of Medical Physiology
Clinical Correlations
| Drug/Condition | Mechanism | Calcium Effect |
|---|---|---|
| Furosemide (loop diuretic) | Blocks NKCC2 → removes lumen+ voltage in TAL | ↑ Ca²⁺ excretion (hypercalciuria) → treats hypercalcaemia |
| Thiazides | Block NCC in DCT → ↑ NCX1 activity | ↓ Ca²⁺ excretion (hypocalciuria) → treats kidney stones |
| Hyperparathyroidism | ↑ PTH → ↑ reabsorption at TAL + DCT | ↑ serum Ca²⁺ (also ↑ gut absorption via calcitriol) |
| Hypoparathyroidism | ↓ PTH → ↓ reabsorption | ↓ serum Ca²⁺, hypercalciuria |
| Volume depletion | ↑ PCT Na⁺/H₂O reabsorption (passive drag) | ↑ Ca²⁺ reabsorption at PCT |
Inhibition of hepatic gluconeogenesis
Checking Date & Time
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
This question is almost certainly about metformin, the first-line drug for type 2 diabetes (which this patient has). Here is the complete explanation:
Inhibition of Hepatic Gluconeogenesis — Metformin's Primary Mechanism
Why Does This Matter for This Patient?
In type 2 diabetes, the liver is insulin-resistant. Normally insulin suppresses gluconeogenesis, but because the liver ignores insulin's signal, it keeps making new glucose from non-sugar substrates (amino acids, lactate, glycerol) even in the fed state — especially overnight. This drives fasting hyperglycaemia. Metformin directly targets this problem.
The Normal (Pathological) State — Why Gluconeogenesis is Overactive in T2DM
Glucagon (or insulin resistance)
↓
↑ cAMP in hepatocyte
↓
CREB (transcription factor) activated
↓
CREB + TORC2 (coactivator) → enter nucleus together
↓
↑ Transcription of PGC1α (peroxisome proliferator-activated receptor-γ coactivator 1α)
↓
PGC1α activates key gluconeogenic enzymes:
• PEPCK (phosphoenolpyruvate carboxykinase)
• Glucose-6-phosphatase (G6Pase)
↓
↑ Gluconeogenesis → ↑ Glucose export → ↑ Fasting blood glucose
In T2DM, this pathway remains continuously stimulated even when insulin is present, because the liver is resistant to insulin's suppressive effects.
How Metformin Blocks This
Metformin → activates LKB1 (upstream kinase) → activates AMPK → phosphorylates TORC2 → sequesters TORC2 in the cytoplasm → CREB cannot activate PGC1α → ↓ gluconeogenic enzyme transcription → ↓ hepatic glucose output
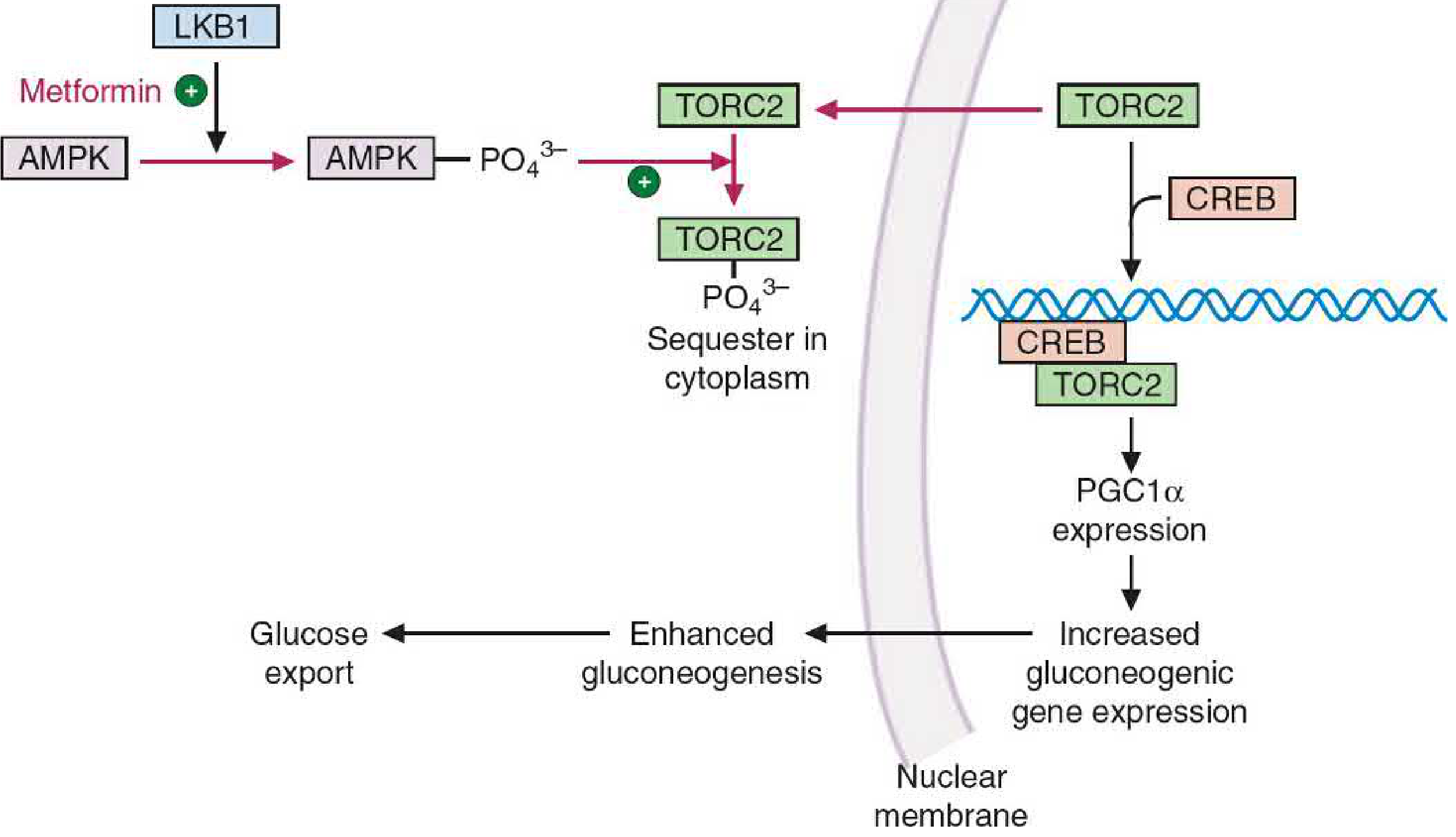
Step-by-Step Molecular Mechanism
| Step | What Happens |
|---|---|
| 1 | Metformin enters hepatocytes via OCT1 (organic cation transporter) |
| 2 | Metformin activates LKB1 (serine/threonine kinase) via unknown mechanism |
| 3 | LKB1 phosphorylates and activates AMPK (AMP-activated protein kinase) |
| 4 | Active AMPK phosphorylates TORC2 (transducer of regulated CREB activity 2) |
| 5 | Phospho-TORC2 is sequestered in the cytoplasm — cannot enter the nucleus |
| 6 | Without TORC2, CREB is inefficient at transcribing the PGC1α gene |
| 7 | ↓ PGC1α → ↓ expression of PEPCK and Glucose-6-phosphatase |
| 8 | ↓ Gluconeogenesis → ↓ hepatic glucose export → ↓ fasting blood glucose |
— Basic Medical Biochemistry, 6th ed Lippincott, p. 1225–1226
Additional Actions of Metformin (Beyond Gluconeogenesis)
AMPK activation has multiple beneficial downstream effects:
| Effect | Mechanism |
|---|---|
| ↓ Lipogenesis | AMPK inhibits acetyl-CoA carboxylase → ↓ malonyl-CoA → ↓ fatty acid synthesis; AMPK also inhibits SREBP-1 transcription → ↓ lipogenic enzymes |
| ↑ Peripheral glucose uptake | AMPK activates GLUT4 translocation in skeletal muscle |
| ↓ Intestinal glucose absorption | Slows carbohydrate absorption from gut |
| ↓ Cholesterol synthesis | AMPK phosphorylates and inhibits HMG-CoA reductase (same enzyme statins target!) |
Key Clinical Points
| Feature | Detail |
|---|---|
| Does NOT cause hypoglycaemia | Does not stimulate insulin secretion; lowers glucose only when liver is overproducing it |
| Preferred 1st-line drug for T2DM | Including this patient |
| Weight neutral/promotes weight loss | Reduces appetite |
| Lactic acidosis risk | Metformin inhibits mitochondrial complex I → ↑ lactate; risk ↑ with renal impairment (eGFR <30 → contraindicated) |
| Vitamin B12 deficiency | Long-term use impairs B12 absorption in terminal ileum |
| Hold before contrast | Risk of contrast nephropathy → ↑ lactate accumulation |
Connecting back to this patient: He has T2DM and just had a PCI — metformin is ideal (cardiovascular neutral to beneficial), but should have been held around the time of his PCI due to contrast dye use, and requires monitoring of renal function (eGFR) given his cardiovascular disease.
— Lippincott Illustrated Reviews Pharmacology, p. 811–812
This is a shared conversation. Sign in to Orris to start your own chat.