Teach me hemolytic anemia in simple way for pathology University examin very simple language so I can score more
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Loading Image
I have all the information needed for a comprehensive, exam-focused answer. Let me now compose a well-structured, easy-to-understand guide.
🩸 Hemolytic Anemia - Simple Pathology Guide for Exam
Based on Robbins & Kumar Basic Pathology (Robbins Pathology)
What is Hemolytic Anemia? (The Big Idea)
Think of it this way: Normal RBCs live for 120 days. In hemolytic anemia, they die early - sometimes in just 20 days.
When RBCs break down faster than the bone marrow can make them, you get anemia. The body fights back by:
- Making more RBCs (reticulocytosis)
- Bone marrow expands (erythroid hyperplasia)
- In severe cases, liver + spleen start making blood cells (extramedullary hematopoiesis)
Classification - The Most Important Table to Remember
| Basis | Types |
|---|---|
| Where the defect is | Intrinsic (inside the RBC) vs. Extrinsic (outside the RBC) |
| Where RBC breaks | Extravascular (in spleen) vs. Intravascular (inside blood vessels) |
Extravascular vs. Intravascular Hemolysis
This is a high-yield comparison question in exams!
| Feature | Extravascular | Intravascular |
|---|---|---|
| Where it happens | Spleen (macrophages eat damaged RBCs) | Inside blood vessels (RBCs burst) |
| Cause | RBC can't pass through spleen (less flexible) | Severe membrane damage (complement, toxins, trauma) |
| Jaundice | Yes (bilirubin from macrophage breakdown) | Less prominent |
| Splenomegaly | Yes (spleen works overtime) | Less prominent |
| Hemoglobinuria | No | Yes (Hb spills into urine - urine turns dark) |
| Hemosiderinuria | No | Yes |
| Iron deficiency | No (iron recycled by macrophages) | Possible (iron lost in urine) |
| Gallstones (pigment) | Yes (long-standing cases) | No |
| Haptoglobin | Low (seen in BOTH types) | Low |
Exam tip: Low haptoglobin is a feature of BOTH types of hemolysis.
Major Hemolytic Anemias - One by One
1. Hereditary Spherocytosis (HS)
Genetics: Autosomal Dominant (mostly)
The Problem (Simple explanation):
The RBC membrane has a "scaffold" made of proteins - Spectrin, Ankyrin, Band 3, Band 4.1, Band 4.2. If any of these are defective, the membrane becomes unstable. The RBC sheds small pieces of membrane, shrinks, and becomes a sphere (spherocyte) instead of a biconcave disc.
Spherocytes are rigid and can't squeeze through the spleen's narrow passages, so they get eaten by macrophages there.
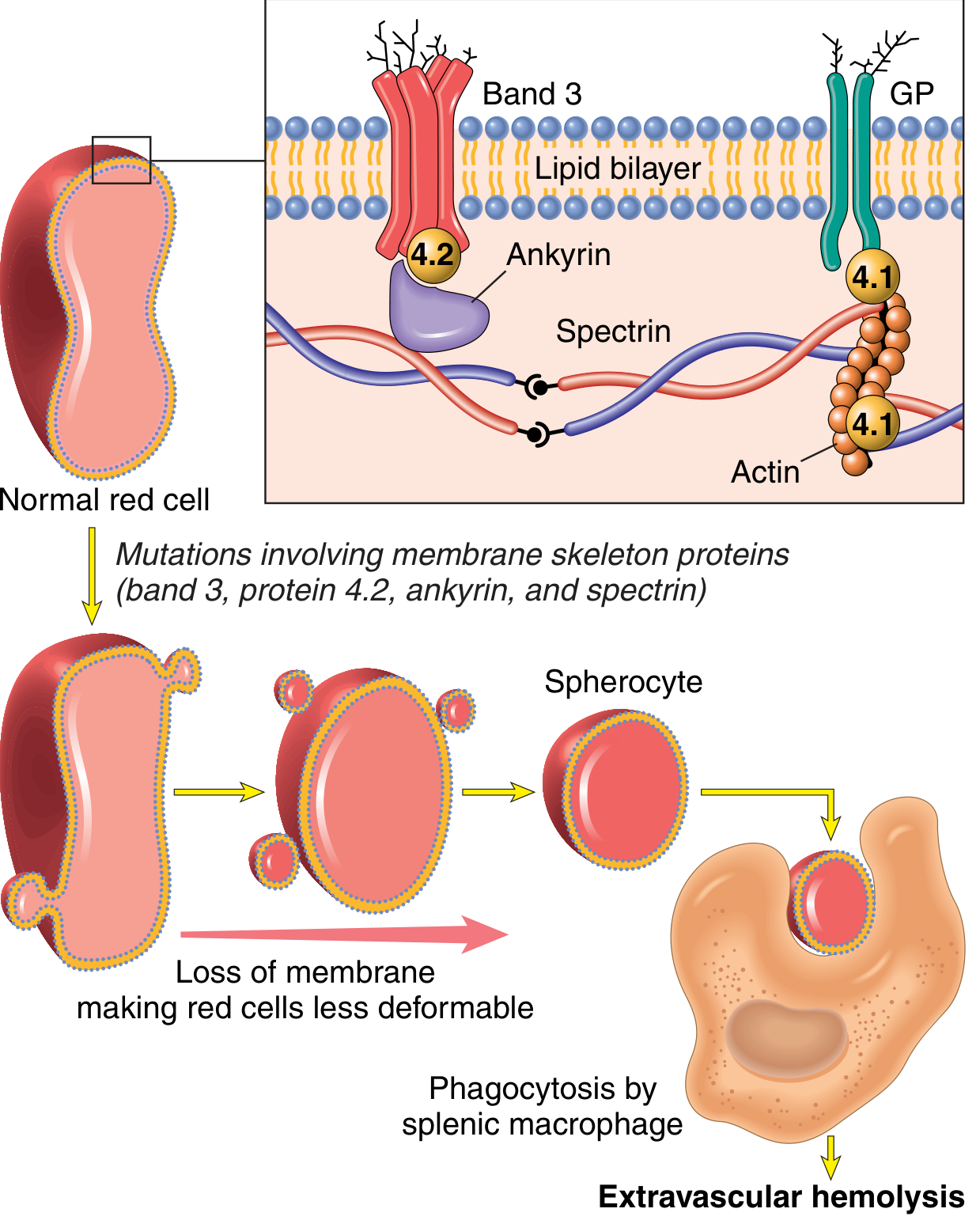
Key proteins mutated: Spectrin, Ankyrin, Band 3, Band 4.2
Type of hemolysis: Extravascular (in spleen)
Clinical features (the triad):
- Anemia
- Splenomegaly (spleen weight 500-1000g; normal is 150-200g)
- Jaundice + Gallstones (pigment stones in 40-50%)
Blood smear: Spherocytes = dark red cells with NO central pallor
Lab findings: Reticulocytosis, elevated indirect bilirubin, low haptoglobin, positive osmotic fragility test
Treatment: Splenectomy - fixes the anemia even though spherocytes remain!
2. Sickle Cell Anemia
Genetics: Autosomal Recessive
The Problem (Simple explanation):
One amino acid swap - Glutamic acid → Valine at position 6 of beta-globin → creates HbS.
When oxygen is low (deoxygenated), HbS molecules stick together and form long polymer rods that distort the RBC into a sickle shape.
Factors that make sickling WORSE:
- Low oxygen
- Dehydration (concentrates HbS)
- Acidosis
- Infection/inflammation (slows blood flow)
- HbF reduces sickling (that's why newborns are protected initially)
Two major consequences:
- Hemolytic anemia - mean RBC life span only 20 days (1/6 of normal)
- Vaso-occlusion - sickled cells block small vessels → pain crises + tissue infarcts
Organs affected:
- Spleen: repeated infarcts → "autosplenectomy" (spleen shrinks and becomes fibrotic) → patient vulnerable to bacterial infections (especially encapsulated bacteria like Pneumococcus)
- Bones: marrow hyperplasia → "crew-cut" appearance on skull X-ray; osteonecrosis of femoral head
- Kidney: papillary necrosis, hematuria
- Brain: stroke
- Eyes: retinopathy
- Lungs: acute chest syndrome
Blood smear: Sickle cells, target cells, anisocytosis, poikilocytosis
Lab: Low Hb, high reticulocytes, elevated bilirubin, Hb electrophoresis shows HbS
3. Thalassemia
Genetics: Autosomal Codominant
The Problem (Simple explanation):
Normal hemoglobin (HbA) = 2 alpha + 2 beta chains. In thalassemia, one type of chain is produced in reduced amounts. The imbalance is the problem.
Beta-Thalassemia Major (Most severe - exam favourite!)
- Both beta-globin genes are mutated → almost no beta chains made
- Alpha chains accumulate → precipitate inside RBC → damage membrane → ineffective erythropoiesis + hemolysis
- Compensatory massive bone marrow expansion → bone deformities
- "Chipmunk face" (maxillary expansion), "hair-on-end" skull X-ray
- Requires regular blood transfusions → iron overload → heart failure, liver cirrhosis, organ damage
- Splenomegaly is massive
Alpha-Thalassemia
- Loss of 1-2 alpha genes: silent carrier to mild anemia
- Loss of 3 alpha genes: HbH disease
- Loss of all 4 alpha genes: Hb Barts → hydrops fetalis (fatal in utero)
Blood smear: Microcytic, hypochromic RBCs, target cells
4. G6PD Deficiency
Genetics: X-linked Recessive (males affected more)
The Problem (Simple explanation):
G6PD enzyme protects RBCs from oxidative damage by making NADPH. Without G6PD, RBCs can't fight oxidative stress → hemoglobin gets oxidized → forms Heinz bodies (denatured Hb clumps) → RBCs destroyed.
Triggers (remember these for exams!):
- Drugs: Primaquine, Dapsone, Nitrofurantoin, Sulfonamides
- Infection
- Fava beans ("Favism")
Type of hemolysis: Episodic (only during triggers, not constant)
Blood smear during crisis: Heinz bodies (on special stain), bite cells (macrophages bite out Heinz body pieces)
Key point: Between attacks, blood smear is NORMAL.
5. Autoimmune Hemolytic Anemia (AIHA)
The Problem: Body makes antibodies against its own RBCs.
| Feature | Warm AIHA | Cold AIHA |
|---|---|---|
| Antibody type | IgG | IgM |
| Temperature | Binds at 37°C (body temp) | Binds at cold temps |
| Hemolysis type | Extravascular (spleen) | Intravascular |
| Causes | SLE, CLL, drugs | Mycoplasma infection, EBV (mono) |
| Treatment | Steroids | Avoid cold |
Diagnosis: Direct Coombs test (DAT) - positive. RBCs are coated with antibody.
6. Microangiopathic Hemolytic Anemia (MAHA) - Bonus!
The Problem: RBCs get shredded as they pass through damaged or partially blocked small vessels (mechanical fragmentation).
Causes: TTP, HUS, DIC, malignant hypertension, artificial heart valves
Blood smear: Schistocytes (helmet cells, fragmented RBCs) - this is pathognomonic!
High-Yield Lab Pattern for ALL Hemolytic Anemias
| Test | Result | Why |
|---|---|---|
| Hemoglobin | Low | RBCs destroyed |
| Reticulocytes | HIGH | Bone marrow compensating |
| Indirect (unconjugated) bilirubin | HIGH | Hb breakdown product |
| Haptoglobin | LOW | Binds free Hb, then removed from circulation |
| LDH | HIGH | Released from broken RBCs |
| Peripheral smear | Abnormal RBC shapes | Key to diagnosis |
Quick Memory Summary
| Disease | Key Defect | Key Finding |
|---|---|---|
| Hereditary Spherocytosis | Spectrin/Ankyrin mutation | Spherocytes, no central pallor |
| Sickle Cell | HbS (Glu→Val) | Sickle cells, vaso-occlusion |
| Thalassemia | Reduced globin chain synthesis | Microcytic hypochromic, target cells |
| G6PD Deficiency | Oxidant sensitivity | Heinz bodies, bite cells, triggered by drugs |
| Warm AIHA | IgG antibody | + Coombs test, responds to steroids |
| Cold AIHA | IgM antibody | + Coombs, triggered by cold, mycoplasma |
| MAHA | Mechanical fragmentation | Schistocytes |
Common Exam Questions & Answers
Q: What is the hallmark of ALL hemolytic anemias?
A: Reticulocytosis (bone marrow compensation) + low haptoglobin.
Q: Why does splenectomy cure hereditary spherocytosis?
A: The spleen is the site of RBC destruction. Remove the spleen → no destruction → anemia corrects. Spherocytes still exist but are no longer destroyed.
Q: What is the most common cause of death in thalassemia major?
A: Cardiac failure from iron overload (hemosiderosis from repeated transfusions).
Q: What blood smear finding is pathognomonic of MAHA?
A: Schistocytes (fragmented RBCs).
Q: Sickle cell patients get infections with which organisms?
A: Encapsulated bacteria - Streptococcus pneumoniae, H. influenzae, Salmonella (especially for osteomyelitis).
Source: Robbins & Kumar Basic Pathology (Robbins Pathology), Chapter 10
This is a shared conversation. Sign in to Orris to start your own chat.