Homogeneous hb E
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Here is a detailed answer:
Homogeneous Hb E (Hb EE Disease)
Molecular Basis
Hb E results from a point mutation at codon 26 of the beta-globin gene - glutamic acid is replaced by lysine (p.Glu26Lys, β26 Glu→Lys). This missense variant does more than change the amino acid: it activates a cryptic donor splice site in codon 25 that competes with the normal donor site. A moderate proportion of RNA is processed through this alternative pathway, reducing the rate of synthesis of the abnormal β-chain. This is why Hb E behaves like a mild β+ thalassemia at the molecular level, even though it produces a structurally abnormal hemoglobin.
- Thompson & Thompson Genetics and Genomics in Medicine, 9th ed., p. 263
Epidemiology
Hb E is probably the most common structurally abnormal hemoglobin in the world, with particularly high frequency in Southeast Asia (Thailand, Laos, Cambodia, Southern China) and the Indian subcontinent. Frequencies approach 60% in parts of Thailand, Laos, and Cambodia. Globally there are estimated to be at least 1 million homozygotes and 30 million heterozygotes.
- Goldman-Cecil Medicine, p. 1710
- Thompson & Thompson Genetics and Genomics in Medicine, 9th ed.
Genetics: Homozygous State (Hb EE)
| Feature | Homozygous Hb E (E/E) |
|---|---|
| Genotype notation | β^E β^E |
| Hb E fraction | ~94% (>90%) |
| Hb A fraction | <4% |
| Hb F | <2% (within or marginally above reference) |
| Hb A2 | Coelutes with Hb E on HPLC |
In homozygous Hb E, no normal β-globin chains are produced, so virtually no Hb A is present. Hb E (α₂β₂^E) predominates, with a small amount of Hb F.
Clinical Features
Homozygous Hb E individuals are usually asymptomatic, despite thalassemic-type CBC indices:
- Normal to mildly decreased hemoglobin (slight anemia at most)
- Low MCV and MCH (microcytic, hypochromic picture)
- Occasional mild splenomegaly
- Normal iron studies
- No jaundice, bone changes, or hepatomegaly
This is a key distinguishing point - the clinical picture is remarkably benign for what appears to be a significant hemoglobinopathy.
- Goldman-Cecil Medicine: "Homozygous HbEE individuals have mild anemia and microcytosis, occasional splenomegaly."
CBC and Peripheral Smear
-
Hemoglobin: Normal or slightly low
-
MCV: Markedly decreased (↓↓)
-
MCH: Low
-
RBC count: Usually elevated (as in thalassemia)
-
Peripheral smear: Numerous target cells, microcytosis, hypochromia
-
This pattern resembles thalassemia trait - hence Hb E is sometimes called a "thalassemic variant"
-
Tietz Textbook of Laboratory Medicine, 7th ed.
-
Quick Compendium of Clinical Pathology, 5th ed.
Laboratory Diagnosis
HPLC
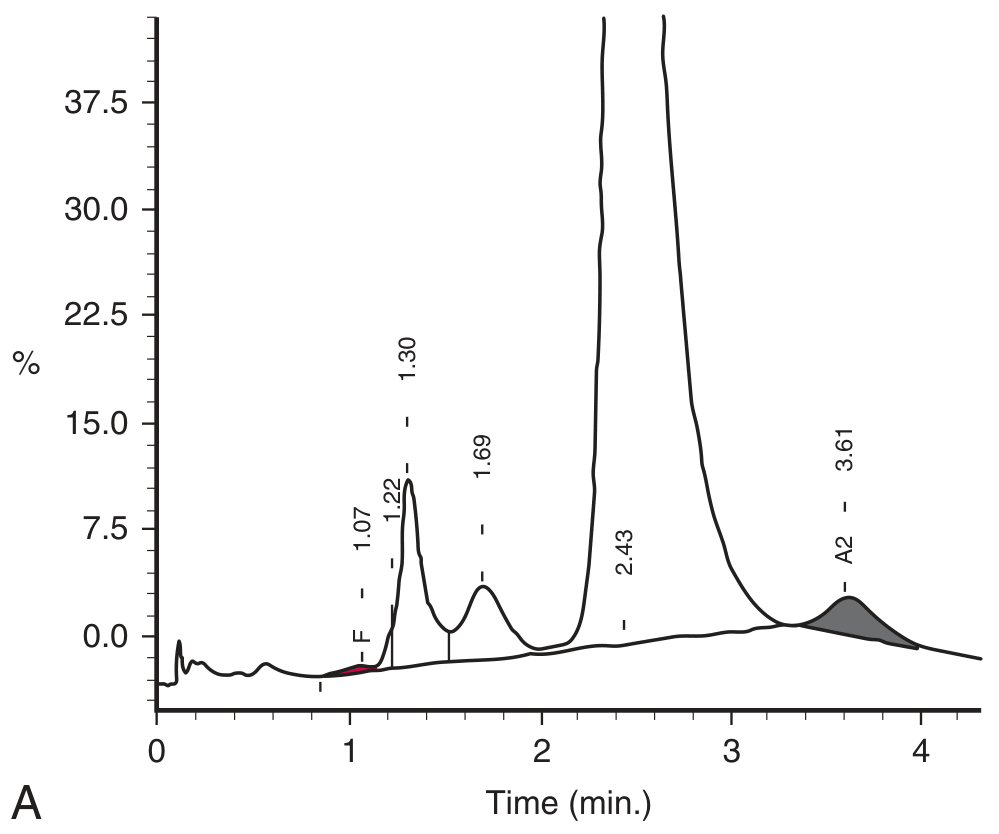
- Single dominant peak (>90%) co-eluting at the Hb A2 position (~3.6 min retention time)
- Hb F within or just above reference range
- Hb E and Hb A2 cannot be separated by HPLC - they co-elute, making accurate Hb A2 quantification impossible
- Capillary zone electrophoresis (CZE) is required to separate Hb E from Hb A2
HPLC trace showing homozygous Hb E - the dominant peak at the A2 window:
Hemoglobin Electrophoresis
- Alkaline electrophoresis: Single band in the C position (same as Hb C, Hb OArab)
- Acid electrophoresis: Migrates to the A position (this distinguishes it from Hb C and Hb OArab, which remain at C on acid gel)
Why Hb A2 is elevated in Hb E
Reduced synthesis of the abnormal β^E chain → excess α-globin chains have more opportunity to bind δ-globin chains → increased Hb A2 production. However, since Hb E co-elutes with Hb A2 on HPLC, the apparent Hb A2 is artefactually very high.
- Tietz Textbook of Laboratory Medicine, 7th ed.
Genotype-Phenotype Summary Table (from Creasy & Resnik)
| Genotype | Anemia | MCV | % A+A₂ | % E | % F | Phenotype |
|---|---|---|---|---|---|---|
| A/E (trait) | None | ↓ | 68 | 30 | <2 | None |
| E/E (homozygous) | 0 to + | ↓↓ | <4 | 94 | <2 | None |
| E/α-Thal | + to ++ | ↓ | 50 | 15 | 35 | None |
| S/E | ++ | ↓ | 0 | 40 | - | None |
| E/β⁺-Thal | ++ | ↓↓ | 10 | 60 | 30 | Splenomegaly |
| E/β⁰-Thal | +++ | ↓↓ | 0 | 60 | 40 | Splenomegaly |
- Creasy & Resnik's Maternal-Fetal Medicine, Table 55.5
Iron Metabolism
An important but clinically underrecognized feature: hepcidin is suppressed in Hb EE, leading to iron accumulation even without blood transfusions. This worsens with transfusions if given unnecessarily.
- Goldman-Cecil Medicine
Interactions and Compound Heterozygotes
Hb E disease itself is benign. However, when Hb E is inherited with a β-thalassemia allele, the combined condition (Hb E/β-thalassemia) becomes the most common severe form of β-thalassemia in Asia, representing about 50% of clinically severe β-thalassemias globally. These patients may be transfusion-dependent. Coinheritance with α-thalassemia produces intermediate severity.
Management
Homozygous Hb E (Hb EE) itself requires no treatment. Patients should be:
- Counseled genetically (risk of severe disease in offspring if partner has β-thalassemia)
- Monitored for iron overload (particularly if any transfusions are given)
- Not given unnecessary iron supplementation (risk of iron loading)
Key summary: Homozygous Hb E is a clinically mild condition - essentially asymptomatic - despite a thalassemic CBC picture with marked microcytosis, low MCH, and numerous target cells. The dominant hemoglobin fraction on HPLC appears at the A2 window. The importance of the diagnosis lies largely in genetic counseling, as coinheritance with β-thalassemia produces a severe, transfusion-dependent disorder.
Management
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
I now have comprehensive material. Here is the full management overview:
Management of Homogeneous Hb E (Hb EE Disease)
Core Principle
Homozygous Hb E is classified as a non-transfusion-dependent thalassemia (NTDT) - the mildest end of the spectrum. Most patients require no active treatment. Management is largely preventive, supportive, and genetic.
1. Reassurance - No Routine Treatment Needed
-
Hb EE individuals are usually asymptomatic with normal or near-normal hemoglobin
-
No regular blood transfusions are required
-
No iron supplementation should be given (see iron section below)
-
Normal life expectancy and quality of life
-
Goldman-Cecil Medicine, p. 1711
2. Iron Management (Critical)
This is the most important active concern in Hb EE:
- Hepcidin is suppressed in Hb EE → increased gastrointestinal iron absorption → iron accumulates even without any transfusions
- Iron supplementation is contraindicated unless true iron deficiency is confirmed (serum ferritin, transferrin saturation)
- If thalassemic red cell indices are misinterpreted as iron deficiency anemia and iron is given inappropriately, iron loading worsens
- Monitor iron stores (ferritin, transferrin saturation) periodically
- If iron overload develops: oral iron chelation with deferasirox or deferiprone; intravenous deferoxamine is an alternative
"In this condition, hepcidin is suppressed, thereby leading to iron accumulation - even in the absence of blood transfusions and certainly worsened by them." - Goldman-Cecil Medicine
3. Folic Acid Supplementation
- Folic acid 2-5 mg/day is generally recommended, especially in:
- Children
- Pregnant women
- Patients with any ongoing hemolysis
- Increased erythropoiesis (even ineffective) raises folate demand
- Goldman-Cecil Medicine; Mulholland's Surgery
4. Blood Transfusion - When to Consider
Hb EE does NOT need regular transfusions, but situational transfusions may be needed in:
| Situation | Rationale |
|---|---|
| Pregnancy (2nd/3rd trimester) | Increased demand may worsen anemia |
| Acute infection/illness | Hemolytic crisis with intercurrent illness |
| Surgery | Perioperative hemoglobin optimization |
| Symptomatic anemia (Hb <7 g/dL) | Uncommon but possible |
Transfusion guidelines when required:
-
Use leukoreduced packed red cells to minimize alloimmunization and reactions
-
Maintain pre-transfusion Hb >9-10.5 g/dL if regular transfusions become necessary
-
Avoid unnecessary transfusions - each unit worsens iron loading
-
Goldman-Cecil Medicine; Harrison's Principles of Internal Medicine 22e
5. Splenomegaly / Splenectomy
-
Occasional mild splenomegaly may develop in Hb EE
-
Splenectomy is rarely needed but considered when:
- Symptomatic splenomegaly or hypersplenism
- Transfusion requirements are increasing due to hypersplenism
- The patient cannot receive transfusions or chelation
-
Caution: Splenectomy in NTDT confers a hypercoagulable state - increased risk of thrombosis (portal vein, pulmonary)
-
Post-splenectomy: vaccinate against encapsulated organisms (pneumococcus, meningococcus, Haemophilus influenzae) and give prophylactic penicillin
-
Mulholland & Greenfield's Surgery, 7th ed.
-
Miller's Anesthesia, 10th ed.
6. Newer / Emerging Therapies (Applicable if Hb E/β-thal or Severe NTDT)
These are not needed for simple Hb EE but become relevant if there is coinheritance:
Luspatercept
- Recombinant fusion protein - binds TGF-β superfamily ligands, reduces Smad2/3 signaling → enhances late-stage erythropoiesis
- Approved for transfusion-dependent β-thalassemia in adults
- Dose: 1.0-1.25 mg/kg SC every 3 weeks
- Reduces transfusion requirements by ~33%
- Goldman-Cecil Medicine; Harrison's 22e
Hydroxyurea
- Increases HbF synthesis (γ-globin induction)
- Benefit in β-thalassemia is limited compared to sickle cell disease (higher HbF levels needed for effect in thalassemia)
- May have a role in Hb E/β-thalassemia, not in simple Hb EE
Hematopoietic Stem Cell Transplantation (HSCT)
- Only curative option for severe hemoglobinopathies
- Not indicated for homozygous Hb EE (disease too mild)
- Relevant only for Hb E/β⁰-thalassemia patients with severe, transfusion-dependent disease
- Requires HLA-matched donor; best results in young patients before iron overload develops
Gene Therapy
-
Lentiviral vectors and CRISPR/Cas9 approaches targeting β-globin locus or HbF induction
-
Approved therapies (e.g., betibeglogene autotemcel) now available for non-β⁰/β⁰ genotype β-thalassemia
-
Not applicable to homozygous Hb EE
-
Goldman-Cecil Medicine; Emery's Elements of Medical Genetics
7. Genetic Counseling (High Priority)
This is arguably the most important management step for Hb EE patients:
-
The condition itself is benign, but offspring are at risk if the partner carries β-thalassemia
-
Hb E/β⁰-thalassemia = severe, transfusion-dependent disease (50% of severe β-thalassemia globally)
-
Partner testing for β-thalassemia trait is mandatory
-
If both partners are carriers, offer prenatal diagnosis:
- Chorionic villus sampling (CVS) at 10-12 weeks
- Amniocentesis at 16 weeks
- DNA analysis for both β-globin gene mutations
-
Thalassemia services should ideally be managed in specialized multidisciplinary centers
-
Goldman-Cecil Medicine; Harrison's 22e; Creasy & Resnik's Maternal-Fetal Medicine
8. Monitoring Schedule (NTDT Guidelines)
| Parameter | Frequency |
|---|---|
| CBC + reticulocytes | Annually (more often if symptomatic) |
| Serum ferritin | Every 6-12 months |
| Transferrin saturation | Every 6-12 months |
| Liver function tests | Annually |
| Echocardiogram (if iron overloaded) | As indicated |
| Endocrine function (growth, puberty) | Pediatric patients |
| Ophthalmology / audiology | If on chelation therapy |
Summary Table
| Aspect | Homozygous Hb E (EE) |
|---|---|
| Regular transfusion | Not required |
| Iron supplementation | Contraindicated (unless proven deficient) |
| Iron chelation | If iron overload develops |
| Folic acid | Recommended (2-5 mg/day) |
| Splenectomy | Rare - only for symptomatic hypersplenism |
| HSCT / Gene therapy | Not indicated |
| Genetic counseling | Mandatory - screen partner for β-thalassemia |
| Luspatercept | Not indicated (reserved for TDT) |
This is a shared conversation. Sign in to Orris to start your own chat.