Megakaryocytic thrombocytopenia
Reading File
Finding Sources
Finding Sources
Searching PubMed
amegakaryocytic thrombocytopenia MPL mutation
Reading File
Reading File
Reading File
Reading File
Finding Sources
Searching PubMed
congenital amegakaryocytic thrombocytopenia CAMT treatment
Finding Sources
Loading Image
Reading File
I now have comprehensive information to give a thorough, well-cited answer. Let me compile it.
Amegakaryocytic (Megakaryocytic) Thrombocytopenia
Overview
Megakaryocytic thrombocytopenia refers to a group of conditions in which thrombocytopenia arises from absence or severe reduction of megakaryocytes in the bone marrow - the precursor cells responsible for platelet production. It exists in two major forms: congenital (CAMT) and acquired.
Congenital Amegakaryocytic Thrombocytopenia (CAMT)
Genetics and Pathophysiology
CAMT is an autosomal recessive disorder caused by loss-of-function mutations in the MPL gene (chromosome 1p34), which encodes the thrombopoietin (TPO) receptor. Without functional TPO signaling, megakaryocyte differentiation from hematopoietic stem cells (HSCs) fails at the very first step - the TPO/MPL axis is the primary driver of early megakaryopoiesis (see diagram below).
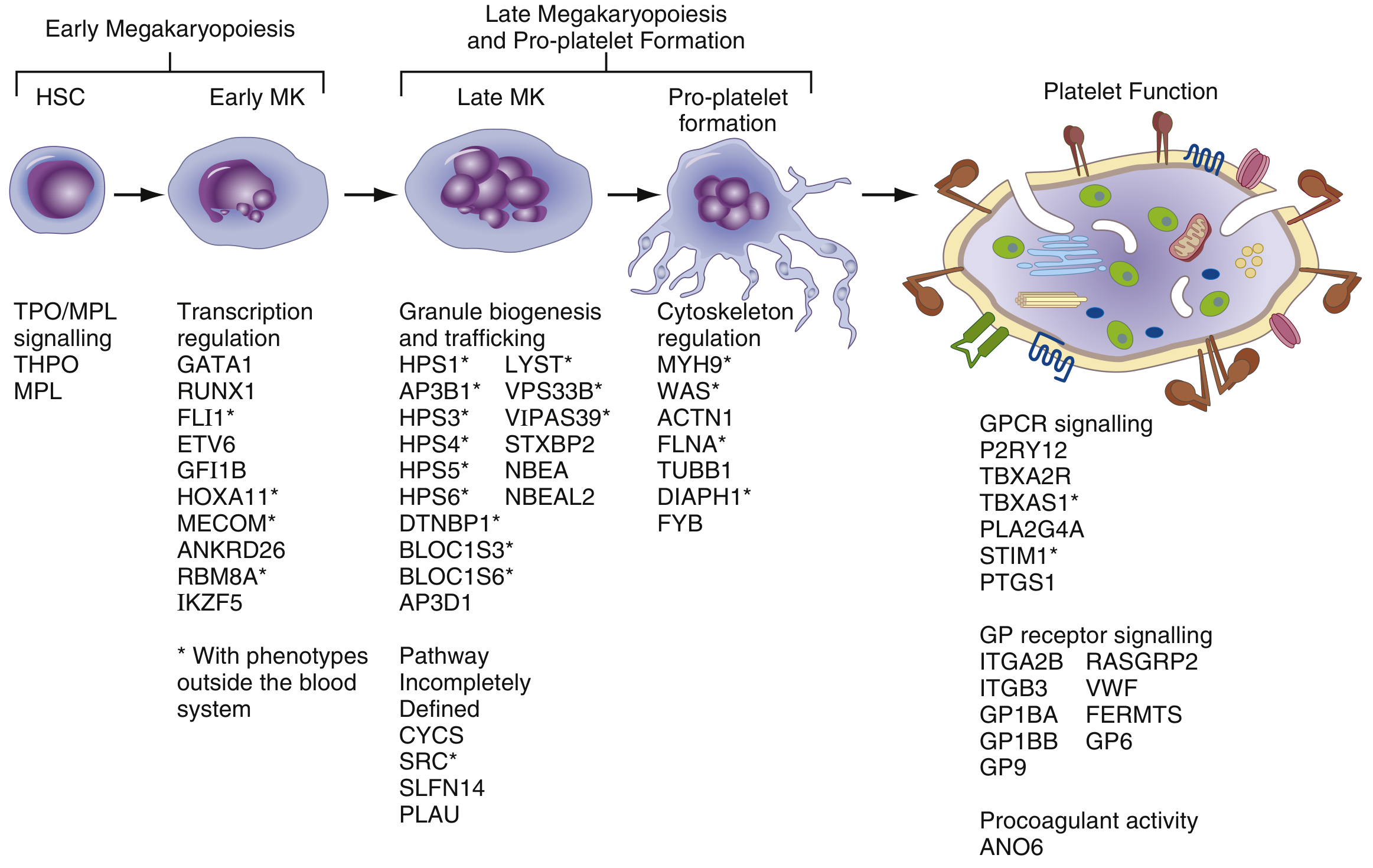
Figure: The megakaryopoiesis pathway - TPO/MPL signaling drives early HSC to early megakaryocyte (MK) differentiation. CAMT results from defects at this earliest stage. (Henry's Clinical Diagnosis and Management, Fig. 41.5)
Clinical Features
| Feature | Details |
|---|---|
| Onset | Neonatal / infancy |
| Presentation | Severe thrombocytopenia with petechiae, purpura, bleeding |
| Bone marrow | Absent or markedly reduced megakaryocytes; otherwise normal (initially) |
| No physical anomalies | Unlike TAR syndrome, no skeletal defects |
| Natural history | Progressive thrombocytopenia → pancytopenia by the second decade (aplastic anemia) |
- Quick Compendium of Clinical Pathology, 5th ed.
CAMT Types
Two subtypes based on the degree of MPL function:
- Type I: Null mutations (nonsense, frameshift) - no residual MPL function; earlier, more severe course with rapid progression to aplastic anemia
- Type II: Missense mutations - some residual receptor function; milder initial course, later progression
Differential Diagnosis
Must be distinguished from other inherited marrow failure syndromes:
| Disorder | Key Distinguishing Feature |
|---|---|
| Thrombocytopenia with absent radii (TAR) | Bilateral radial aplasia; thrombocytopenia most profound at birth but improves; does NOT progress to aplastic anemia |
| Fanconi anemia | Chromosomal fragility, characteristic physical anomalies |
| Dyskeratosis congenita | Skin/nail/mucosal triad, telomere biology mutations |
| Shwachman-Diamond syndrome | Exocrine pancreatic insufficiency, neutropenia |
| Wiskott-Aldrich syndrome | X-linked; small platelets (MPV <7 fL); immunodeficiency |
Platelet size classification is a key diagnostic tool: CAMT produces normal-sized platelets (MPV 7-11 fL), unlike MYH9-related disorders or Bernard-Soulier syndrome (large platelets) or Wiskott-Aldrich (small platelets).
- Henry's Clinical Diagnosis and Management by Laboratory Methods, Table 41.3
Acquired Amegakaryocytic Thrombocytopenia
The acquired form is exceedingly rare and, like pure red cell aplasia (PRCA), appears to be caused by a destructive immune response selectively targeting megakaryocytes or their progenitors. It can be drug/toxin-related or idiopathic. In acquired cases:
- The unaffected lineages (erythroid, myeloid) are quantitatively and qualitatively normal initially
- Progression to pancytopenia or leukemia is unusual (in contrast to CAMT)
- Harrison's Principles of Internal Medicine, 22nd ed.
Diagnosis
- CBC: Isolated severe thrombocytopenia (often <20,000/µL at presentation)
- Peripheral smear: Confirm thrombocytopenia; assess platelet size
- Bone marrow biopsy/aspirate: Hallmark - absent or markedly reduced megakaryocytes with otherwise normal or hypercellular marrow (early CAMT) or progressing to hypoplasia
- TPO level: Markedly elevated (receptor non-functional, so TPO accumulates unbound)
- Genetic testing: MPL gene sequencing confirms CAMT diagnosis
- Chromosomal fragility studies: Exclude Fanconi anemia
Treatment
CAMT
- Hematopoietic stem cell transplantation (HSCT) is the only curative therapy. It corrects the underlying MPL deficiency by providing donor HSCs with functional TPO receptors.
- Matched sibling donor (MSD) preferred; haploidentical transplants have been reported with success in recent case series ([Wang et al., 2022, PMID 36379227])
- Supportive care: Platelet transfusions for bleeding episodes; avoid NSAIDs
- Thrombopoietin receptor agonists (eltrombopag, romiplostim): Theoretically ineffective in classic CAMT (the receptor is absent), but may have limited roles in partial MPL function (Type II) - a 2024 review on TPO-RAs in pediatrics ([PMID 38925157]) discusses their evolving use
Acquired amegakaryocytic thrombocytopenia
- Immunosuppression: Anti-thymocyte globulin (ATG), cyclosporine (analogous to aplastic anemia treatment)
- Remove causative agent if drug/toxin-related
- HSCT in refractory cases
Key Summary Points
- CAMT = autosomal recessive, MPL mutation, neonatal severe thrombocytopenia, absent BM megakaryocytes, normal-sized platelets
- Progressively worsens to pancytopenia/aplastic anemia by adolescence
- TPO levels are elevated (not deficient) - the receptor is broken, not the ligand
- Unlike TAR syndrome, there are no skeletal anomalies and the disorder does not improve with age
- Only cure is allogeneic HSCT
- Acquired form is immune-mediated, rare, and less likely to progress to pancytopenia
Recent evidence note: Novel MPL variants continue to be characterized ([Shah et al., 2023, PMID 37438490]), and a 2025 case series identified THPO promoter mutations as an alternative genetic cause ([PMID 40386912]), suggesting the molecular heterogeneity of CAMT is broader than previously appreciated.
This is a shared conversation. Sign in to Orris to start your own chat.