Heart attack mbbs
Reading File
Finding Sources
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND "management"[MeSH Terms]
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
I now have comprehensive textbook content and images. Let me compile the full MBBS-level answer on Myocardial Infarction (Heart Attack):
Myocardial Infarction (Heart Attack) - MBBS Notes
1. Definition
Myocardial infarction (MI) is necrosis of cardiac muscle resulting from ischemia. The 2018 joint task force (US and European Cardiology) defines it as "acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
- Robbins & Kumar Basic Pathology, p. 353
2. Epidemiology & Risk Factors
- MI can occur at virtually any age; frequency rises with age
- ~10% occur before age 40; ~45% before age 65
- Men are at greater risk, but the gap narrows with age
- Women are relatively protected during reproductive years; risk rises post-menopause (declining estrogen)
- Risk factors: Atherosclerosis (main cause), hypertension, diabetes, hyperlipidemia, smoking, obesity, family history
3. Pathogenesis
The vast majority (>90%) are caused by acute coronary thrombosis. The sequence:
- An atheromatous plaque is eroded or disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents
- Platelets adhere, aggregate, and are activated - releasing thromboxane A2, ADP, and serotonin, causing further platelet aggregation and vasospasm
- Coagulation is activated by exposure of tissue factor - adding to the growing thrombus
- Within minutes, the enlarging thrombus completely occludes the coronary lumen
In ~10% of transmural MIs, no occlusive atherosclerosis is found - these are due to vasospasm, embolism (e.g., from AF, valve vegetations), or small vessel disease (vasculitis, amyloid, sickle cell).
- Robbins & Kumar Basic Pathology, pp. 353-354
4. Coronary Artery Territories
| Artery | Territory | Infarct Location |
|---|---|---|
| LAD (most common) | Anterior LV, anterior septum, apex | Anterior MI |
| RCA | Posterior LV, posterior septum, RV, SA/AV nodes | Inferior/posterior MI |
| LCx | Lateral LV | Lateral MI |
5. Types of MI
Based on depth:
- Transmural infarction: Full thickness of ventricle wall; caused by epicardial vessel occlusion with thrombosis → associated with STEMI on ECG
- Subendocardial infarction: Limited to inner third of myocardium; occurs when thrombus is lysed before necrosis becomes transmural, or from global hypoperfusion → associated with NSTEMI
Based on ECG:
- STEMI (ST-elevation MI): Complete occlusion, transmural ischemia
- NSTEMI (Non-ST-elevation MI): Partial occlusion or spontaneous thrombolysis
6. Myocardial Response to Ischemia
- Within seconds: Loss of contractile function (stunning)
- At 20-40 minutes: Irreversible cell death begins in the subendocardium (most vulnerable - farthest from epicardial vessels, highest oxygen demand, compressed during systole)
- Necrosis then spreads outward as a "wavefront" toward epicardium over hours
The heart needs ~1.3 mL O2/100g/min just to survive; normal delivery is ~8 mL O2/100g/min. If 15-30% of normal flow is maintained, muscle may survive.
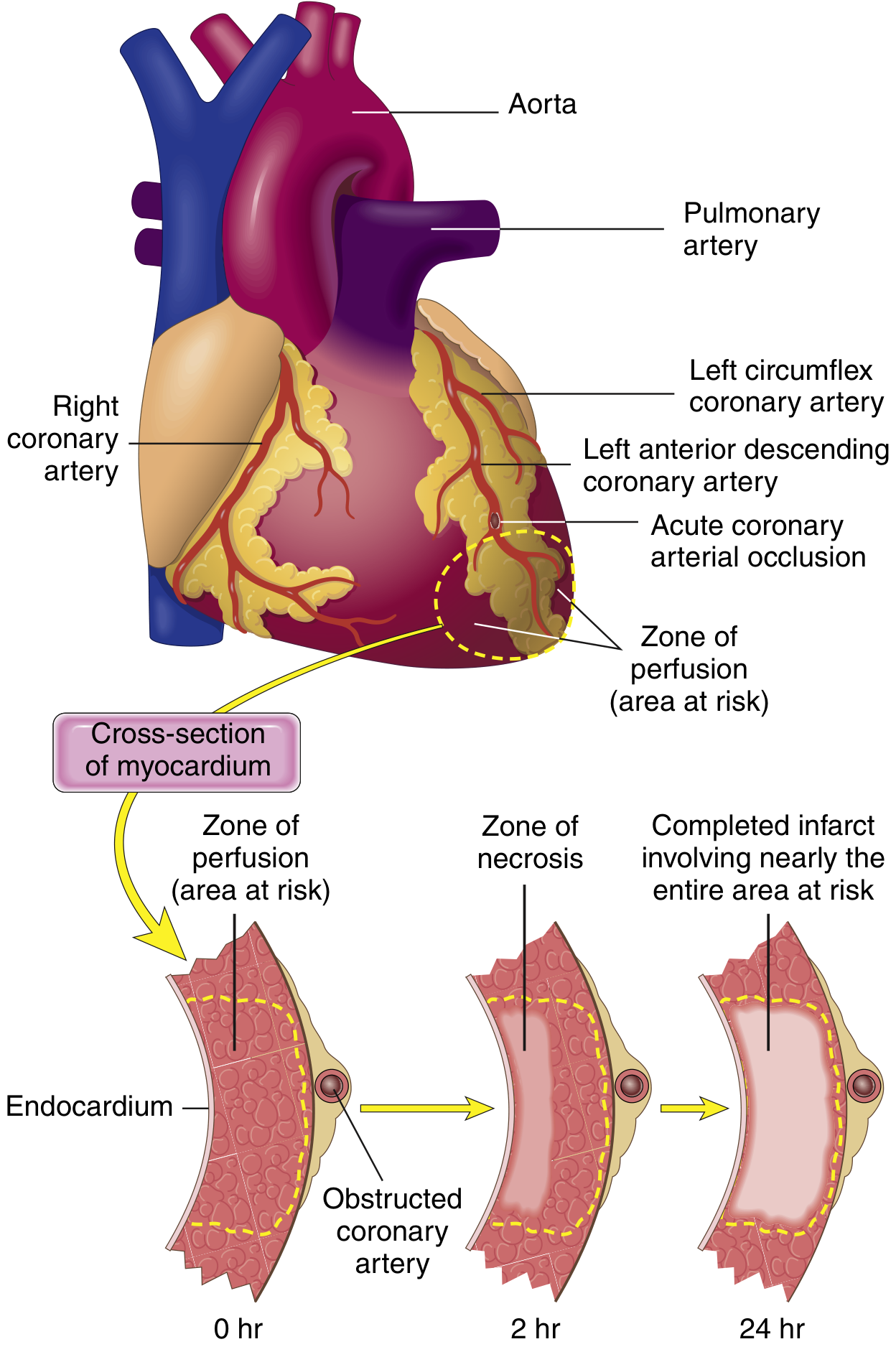
- Guyton & Hall Textbook of Medical Physiology, p. 271
7. Morphology - Timeline (VERY HIGH YIELD)
| Time | Gross Features | Light Microscopy | Key Feature |
|---|---|---|---|
| 0 - 30 min | None | None | Reversible - EM shows mitochondrial swelling, glycogen loss |
| 30 min - 4 hr | None | Usually none; wavy fibers at border | Sarcolemmal disruption (irreversible begins) |
| 4-12 hr | Occasionally dark mottling | Coagulative necrosis begins; edema, hemorrhage | |
| 12-24 hr | Dark mottling | Coagulative necrosis; pyknotic nuclei; hypereosinophilic myocytes; early neutrophils | TTC stain shows pale area |
| 1-3 days | Mottling with yellow-tan center | Full coagulative necrosis; loss of nuclei/striations; peak neutrophils | |
| 3-7 days | Hyperemic border; yellow-tan softening | Dying neutrophils; macrophages begin phagocytosis | Risk of rupture highest |
| 7-10 days | Maximally yellow-tan and soft | Active phagocytosis; early granulation tissue at margins | |
| 10-14 days | Red-gray depressed borders | Well-developed granulation tissue; new vessels; collagen deposition | |
| 2-8 weeks | Gray-white scar | Dense collagenous scar | |
| >2 months | Complete scar | Complete fibrosis |
Key stain: Triphenyl tetrazolium chloride (TTC) - infarcted area appears pale/unstained (LDH leaks out), healthy muscle stains deep red.
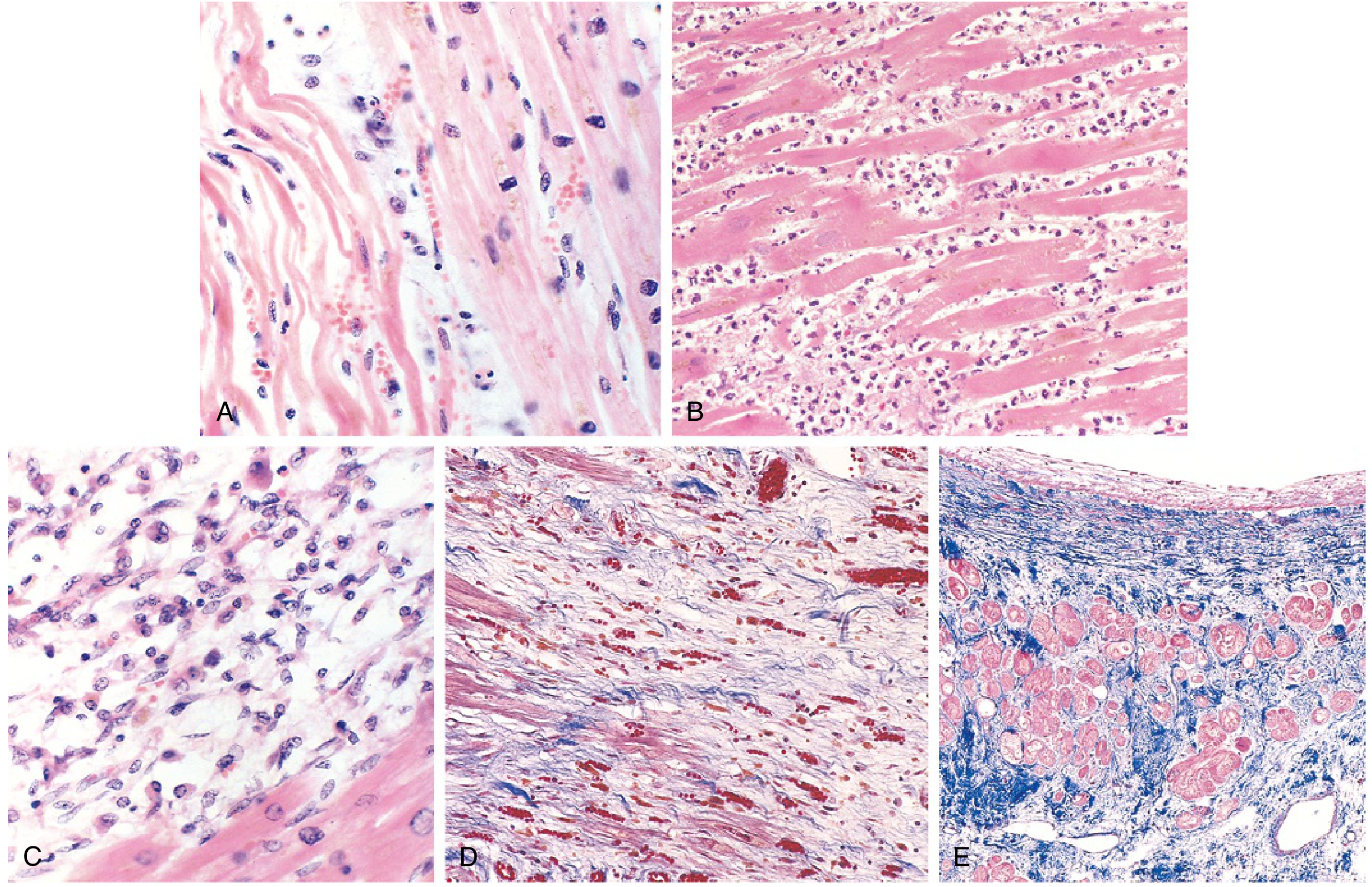
- Robbins & Kumar Basic Pathology, p. 357
8. Clinical Features
Symptoms:
- Severe, crushing/squeezing central chest pain - often radiating to left arm, jaw, neck, back
- Pain lasts >30 minutes (unlike angina which resolves with rest/nitrates)
- Nausea, vomiting, sweating (diaphoresis)
- Dyspnea (from acute LV failure/pulmonary congestion)
- Sense of impending doom
- Silent MI (no pain) - common in diabetics and elderly
Signs:
- Pallor, sweating, clammy skin
- Tachycardia or bradycardia
- Hypotension (cardiogenic shock if extensive)
- S3/S4 gallop
- Mitral regurgitation murmur (papillary muscle ischemia)
9. ECG Changes (High Yield)
Three mechanisms of ECG change (from Ganong's Physiology):
| Defect in Infarcted Cells | Current Flow | ECG Change |
|---|---|---|
| Rapid repolarization (K+ channels open) | Out of infarct | ST elevation |
| Decreased resting membrane potential | Into infarct | TQ depression (appears as ST elevation) |
| Delayed depolarization | Out of infarct | ST elevation |
ECG Evolution:
- Hyperacute T waves - earliest change (minutes to hours)
- ST-segment elevation - classic STEMI, in leads overlying the infarct; reciprocal ST depression in opposite leads
- T-wave inversion - develops hours to days
- Pathological Q waves - develop over days, persist; indicate transmural necrosis (dead silent tissue)
ECG Localization:
-
Anterior MI: V1-V4 (LAD occlusion)
-
Inferior MI: II, III, aVF (RCA occlusion)
-
Lateral MI: I, aVL, V5-V6 (LCx occlusion)
-
Posterior MI: Tall R in V1 (mirror image; use V7-V9)
-
Ganong's Review of Medical Physiology, p. 534
10. Investigations / Diagnosis
Biomarkers (most important):
| Marker | Rises | Peaks | Returns to Normal | Notes |
|---|---|---|---|---|
| Troponin I/T | 3-4 hr | 24-48 hr | 7-14 days | Most sensitive & specific; gold standard |
| CK-MB | 4-6 hr | 18-24 hr | 48-72 hr | Useful for re-infarction detection |
| Myoglobin | 1-2 hr | 6-8 hr | 24 hr | Earliest; not cardiac-specific |
| LDH | 24-48 hr | 3-6 days | 8-14 days | Useful in late presenters |
Other investigations:
- ECG - serial ECGs
- Echocardiography - regional wall motion abnormality
- Coronary angiography - confirms and allows PCI
- Chest X-ray - pulmonary edema, cardiomegaly
- CBC, lipid profile, glucose, renal function
11. Immediate Management ("MONA" + Reperfusion)
Initial stabilization:
- Morphine - analgesia, anxiolysis (use cautiously - may worsen outcomes in some studies)
- Oxygen - if SpO2 <94%
- Nitrates - sublingual GTN for chest pain (contraindicated if hypotensive or RV infarct)
- Aspirin 300 mg loading dose - inhibits platelet aggregation (COX-1 pathway, TXA2)
- P2Y12 inhibitor - Clopidogrel/Ticagrelor (dual antiplatelet therapy)
Reperfusion (time is myocardium):
- Primary PCI (percutaneous coronary intervention) - preferred if available within 90-120 min of first medical contact; stenting + balloon angioplasty
- Thrombolysis (streptokinase, tPA/alteplase) - if PCI not available within 120 min; best given within 12 hours; dissolves thrombus by activating plasminogen
Additional drugs:
- Beta-blockers (metoprolol) - reduce heart rate, O2 demand; reduce arrhythmia risk
- ACE inhibitors - reduce LV remodeling, given within 24 hours
- Statins - high-intensity statin therapy (atorvastatin 80 mg)
- Anticoagulation - Heparin/LMWH/fondaparinux (adjunct to reperfusion)
"Time is myocardium" - Every minute of delay = ~1.9 million myocytes lost.
- Robbins & Kumar Basic Pathology, p. 355
12. Complications
Early (<72 hours):
- Arrhythmias (most common - ~90% develop some rhythm disturbance): VF (most dangerous, highest risk in first hour), VT, heart block, AF
- Cardiogenic shock: When >40% of LV is infarcted; 10% of transmural MIs; high mortality
- Acute LV failure / Pulmonary edema
Day 3-7 (softening phase - danger zone):
- Cardiac rupture (free wall rupture → hemopericardium → tamponade; usually fatal)
- Interventricular septum rupture → acute VSD
- Papillary muscle rupture → acute severe mitral regurgitation
Late (weeks-months):
- Pericarditis - appears 2-3 days post-MI (fibrinous pericarditis), friction rub; Dressler syndrome - autoimmune pericarditis weeks later
- Ventricular aneurysm - thinned, scarred, non-contractile wall (large anterior infarcts); causes mural thrombus, arrhythmia, heart failure; does NOT rupture
- Mural thrombus - risk of systemic embolism
- Progressive heart failure / Ischemic cardiomyopathy
13. Reperfusion Injury
Restoration of blood flow, while beneficial, can cause additional injury:
-
Mitochondrial dysfunction - altered membrane permeability, apoptosis
-
Myocyte hypercontracture - Ca²+ overload → uncontrolled sarcomere contraction → cell death
-
Oxidative stress - ROS (O2-, H2O2, OH·) generated within minutes of reperfusion
-
"No-reflow" phenomenon - leukocyte plugging of microvasculature despite opening the epicardial artery
-
Contraction band necrosis - pathognomonic histologic feature of reperfused myocardium; intense eosinophilic transverse bands of hypercontracted sarcomeres
-
Robbins & Kumar Basic Pathology, p. 358
14. Summary Mnemonics
STEMI management: "TIME"
- Thrombolysis or PCI (reperfusion)
- Infusion of anticoagulant (heparin)
- MONA (Morphine, O2, Nitrates, Aspirin)
- Extras: Beta-blocker, ACE inhibitor, Statin
Causes of early death in MI (Guyton & Hall):
- Decreased cardiac output (cardiogenic shock) - "systolic stretch" of infarcted segments
- Pulmonary edema - damming of blood in pulmonary circulation
- Ventricular fibrillation - most common cause of sudden cardiac death
- Cardiac rupture (rare)
Sources: Robbins & Kumar Basic Pathology (9780323790185); Guyton & Hall Textbook of Medical Physiology (9780443111013); Ganong's Review of Medical Physiology 26th Ed (9781260122404)
This is a shared conversation. Sign in to Orris to start your own chat.