Infectious mononucleosis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"infectious mononucleosis" AND "Epstein-Barr virus"
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a thorough, well-illustrated answer. Here is the comprehensive overview:
Infectious Mononucleosis (IM)
Definition and Etiology
Infectious mononucleosis is an acute, self-limited lymphoproliferative disorder caused by Epstein-Barr virus (EBV), also designated human herpesvirus 4 (HHV-4), a gamma herpesvirus of the Lymphocryptovirus genus. It is responsible for >90% of IM cases. The classic triad is fever, sore throat, and lymphadenopathy, accompanied by atypical lymphocytosis. CMV is the next most common cause and produces a clinically indistinguishable syndrome (heterophile-negative).
- Harrison's Principles of Internal Medicine 22E, p. 1552
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 341
Epidemiology
-
EBV infects >90% of adults worldwide. Transmission is via oral secretions ("kissing disease").
-
In lower-income or low-hygiene settings, primary EBV infection occurs in early childhood and is nearly always asymptomatic.
-
In higher-income settings, infection is delayed to adolescence/young adulthood, and symptomatic IM is far more common (~75% of EBV infections in adolescents manifest as IM).
-
Incubation period: 30-50 days.
-
More than 90% of seropositive individuals shed EBV in oropharyngeal secretions lifelong; shedding is increased during IM and in immunocompromised patients.
-
An estimated 100,000 cases per year occur in the United States.
-
Harrison's 22E, p. 1552; Jawetz, Melnick & Adelberg's Medical Microbiology 28E; Red Book 2021, p. 549
Pathogenesis
EBV is transmitted via saliva. The sequence of events:
- Initial infection of oropharyngeal epithelial cells and salivary glands, with subsequent spread to tonsillar B lymphocytes via the CD21 receptor (also the C3d complement receptor).
- EBV infection of B cells takes two forms:
- Latent (majority): virus persists as an extrachromosomal episome; viral proteins LMP1, LMP2, EBNA2 drive polyclonal B-cell activation and proliferation.
- Lytic (minority): active viral replication, cell lysis, and virion release.
- Latently infected B cells secrete polyclonal antibodies, including heterophile antibodies (react with sheep/horse red cells - the basis of the Monospot test) and autoantibodies (e.g., anti-platelet).
- The host immune response drives symptoms:
- CD8+ cytotoxic T cells (CTLs) and NK cells expand massively to suppress EBV-infected B cells - these are the atypical lymphocytes seen on blood smear.
- During acute IM, up to 1 in 100 B cells in the peripheral blood is EBV-infected; after recovery, this falls to 1-50 in every 1 million B cells.
- There is an inverted CD4+/CD8+ ratio during acute illness; up to 40% of CD8+ T cells target EBV antigens.
- Memory B cells (not epithelial cells) are the long-term reservoir for latent EBV.
- Harrison's 22E, p. 1552-1553; Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar, p. 340
Clinical Manifestations
Classic Triad: Fever + Pharyngitis + Lymphadenopathy
| Manifestation | Frequency |
|---|---|
| Sore throat | 75% (50-87%) |
| Malaise | 47% (42-76%) |
| Headache | 38% (22-67%) |
| Lymphadenopathy | 94% (83-100%) |
| Fever | 76% (60-100%) |
| Pharyngitis/tonsillitis | 74% (50-87%) |
| Splenomegaly | 52% (43-64%) |
| Hepatomegaly | 11% (6-15%) |
| Rash | 5% |
Key clinical points:
-
A prodrome of fatigue, malaise, and myalgia precedes the full picture by 1-2 weeks.
-
Lymphadenopathy is most prominent in the first 2 weeks; posterior cervical nodes are most commonly involved but generalized adenopathy occurs. Nodes are tender, symmetric, and non-fixed.
-
Pharyngitis is often the most prominent sign; tonsils may show exudate resembling streptococcal pharyngitis.
-
Splenomegaly peaks during weeks 2-3.
-
Rash: ~5% spontaneously; historically reported in 70-100% of patients given ampicillin/amoxicillin. More recent studies suggest amoxicillin-associated rashes may not purely be a drug-EBV interaction and are not predictive of future penicillin allergy.
-
Elderly patients: often present atypically with prolonged fever, fatigue, and myalgia; pharyngitis, lymphadenopathy, and atypical lymphocytes are less prominent.
-
Infants/young children: mostly asymptomatic or mild non-specific illness.
-
Illness typically self-resolves in 2-4 weeks; fatigue may persist for months.
-
Harrison's 22E, p. 1553-1554
Pathological Findings (Morphology)
Peripheral blood:
- Absolute lymphocytosis: >60% of WBCs are lymphocytes.
- 5-80% are atypical lymphocytes (Downey cells): large cells 12-16 µm in diameter with abundant vacuolated cytoplasm, oval/indented/folded nucleus, and azurophilic granules. Mostly CD8+ CTLs.
Atypical lymphocytes in IM (Robbins):
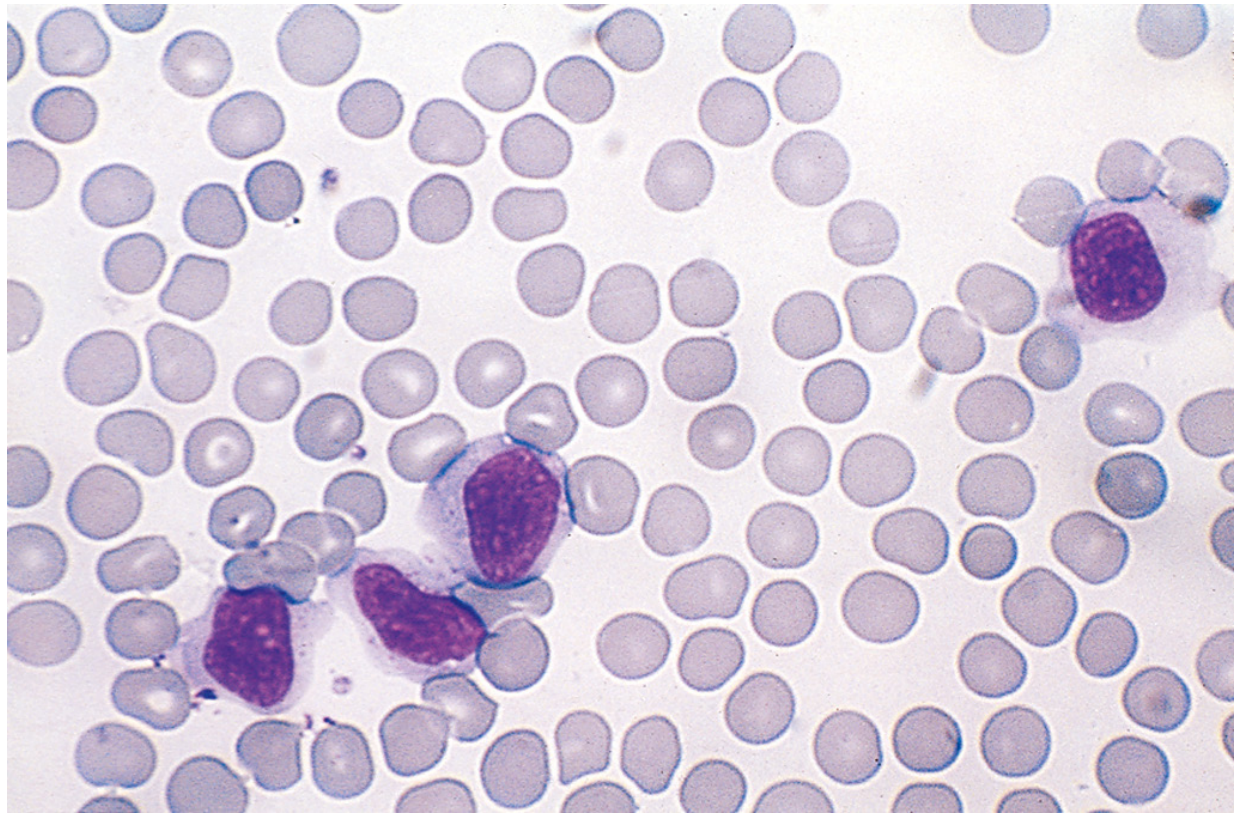
Downey cells on blood smear (Sherris & Ryan):
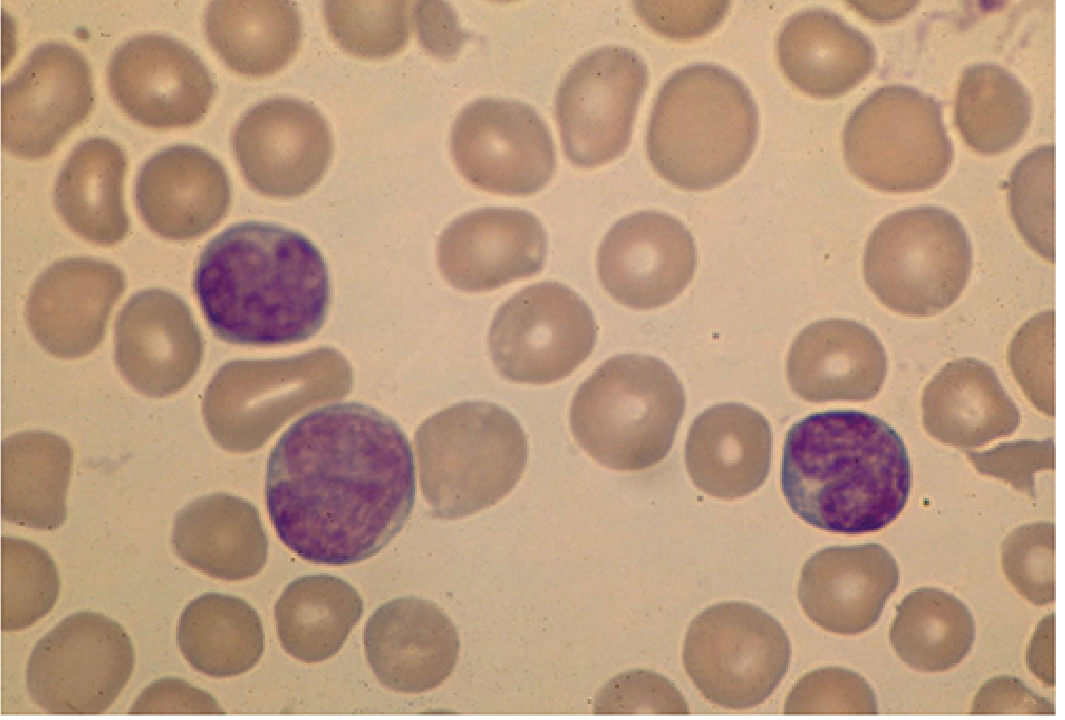
Lymph nodes: Discrete generalized enlargement; paracortical expansion by activated T-cell immunoblasts. EBV-infected B cells may resemble Reed-Sternberg cells - can mimic lymphoma on biopsy.
Spleen: Enlarged (300-500 g), soft, fleshy, hyperemic. The rapid capsular stretching makes it vulnerable to rupture.
Liver: Moderate hepatomegaly; portal and sinusoidal atypical lymphocytic infiltration; focal hepatocyte necrosis (histologically similar to viral hepatitis).
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 341
Diagnosis
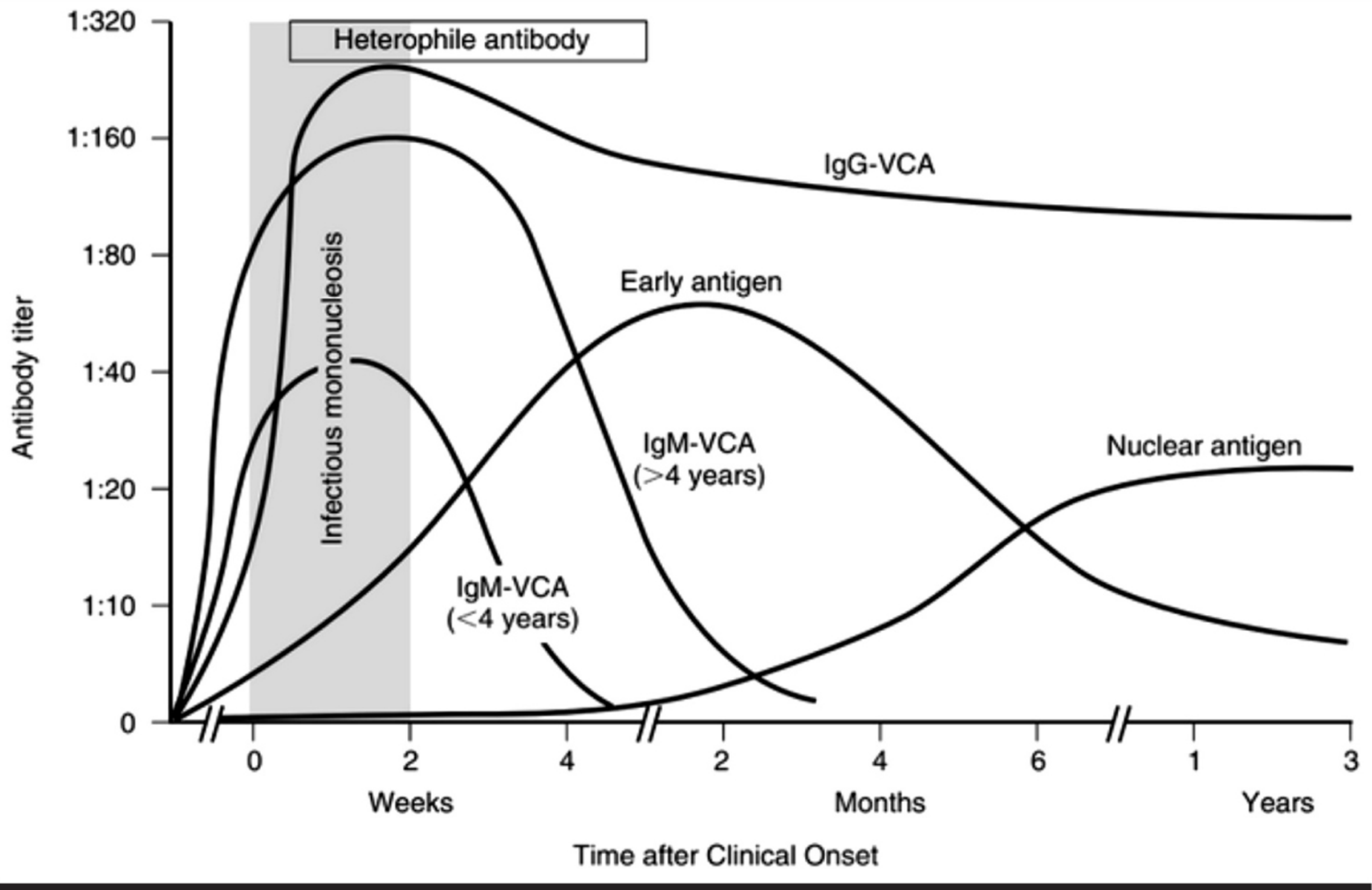
1. Heterophile Antibody (Monospot Test)
- ~90% sensitive and specific in adolescents/adults.
- IgM antibodies; appear in the first 1-2 weeks, peak by week 2-3, disappear within 6 months.
- False negatives: children <4 years (frequently negative); early in illness; in up to 5-15% of adults.
- The test uses animal erythrocytes absorbed with guinea pig kidney (IM heterophile absorbed by sheep RBCs but NOT guinea pig kidney).
2. EBV-Specific Serology
Used when heterophile test is negative, patient is <4 years old, or diagnosis is uncertain.
| EBV Status | VCA IgG | VCA IgM | EA (D) | EBNA |
|---|---|---|---|---|
| No prior infection | - | - | - | - |
| Acute infection | + | + | +/- | - |
| Recent infection | + | +/- | +/- | +/- |
| Past infection | + | - | +/- | + |
Key interpretation rules:
- IgM anti-VCA: present in acute infection, disappears within 4-6 weeks.
- IgG anti-VCA: rises early and persists for life - not useful alone for timing.
- Anti-EBNA (anti-nuclear antigen): ABSENT in acute infection; develops weeks-months after infection and persists lifelong. Its presence effectively rules out acute primary EBV infection.
- Early antigen (EA-D): present transiently; found in about 70% of acute cases.
3. CBC and Differential
- Lymphocytosis with >10% atypical lymphocytes is highly characteristic.
- Lymphocytosis >50% of WBC differential strongly supports the diagnosis.
4. PCR
-
EBV DNA PCR in serum/plasma/tissue; used mainly in immunocompromised patients, complex cases, and for monitoring post-transplant lymphoproliferative disorder (PTLD).
-
Red Book 2021, p. 550; Sherris & Ryan's Medical Microbiology 8E; Harrison's 22E
Differential Diagnosis
| Condition | Key Distinguishing Features |
|---|---|
| CMV mononucleosis | Older age, longer fever, less pharyngitis/lymphadenopathy; heterophile-negative |
| Acute HIV infection | Diffuse rash, oral/genital ulcers, aseptic meningitis |
| Toxoplasmosis | Less splenomegaly; cat exposure; raw meat |
| HHV-6 infection | Older presentation age |
| Streptococcal pharyngitis | No splenomegaly, minimal fatigue, no atypical lymphocytes |
| Viral hepatitis | Higher aminotransferases, no pharyngitis |
| Rubella | Maculopapular rash, no splenomegaly |
| Lymphoma | Fixed, non-tender lymph nodes; constitutional B symptoms |
| Drug reaction (phenytoin, carbamazepine, sulfonamides) | Any age; no atypical lymphocytes typically |
- Harrison's 22E, p. 1555
Complications
Hematologic
- Splenic rupture (most feared; can be spontaneous or from minor trauma) - risk is highest in weeks 2-3.
- Thrombocytopenia (immune-mediated; usually mild)
- Autoimmune hemolytic anemia
- Agranulocytosis
- Hemophagocytic lymphohistiocytosis (HLH)
Neurological
- Aseptic meningitis, encephalitis, transverse myelitis, optic neuritis, cranial nerve palsies, Guillain-Barré syndrome, Alice in Wonderland syndrome (visual perceptual distortions)
Other
-
Airway obstruction (massive tonsillar hypertrophy)
-
Myocarditis, pneumonia, orchitis (rare)
-
Hepatitis (common but usually mild and self-limiting)
-
Chronic fatigue lasting >6 months in ~10% of classic IM cases
-
Red Book 2021, p. 547
Treatment
No specific antiviral therapy is approved or effective for IM. Management is supportive:
- Rest and analgesia - antipyretics/NSAIDs for fever and throat pain.
- Avoid strenuous activity and contact sports for at least 21 days from symptom onset to prevent splenic rupture. Return to contact sports only after 4-7 weeks if asymptomatic with no overt splenomegaly.
- Avoid amoxicillin/ampicillin - causes a non-allergic morbilliform rash in a significant proportion of IM patients. Other penicillins should also be avoided where possible.
- Glucocorticoids - NOT indicated for uncomplicated IM. Used only for specific severe complications:
- Severe tonsillar hypertrophy with impending airway obstruction
- Autoimmune hemolytic anemia
- Severe thrombocytopenia
- HLH
- Myocarditis; severe CNS disease
- Dose: Prednisone 40-60 mg/day (or 1 mg/kg/day in children, max 60 mg/day) for 5-7 days, with tapering.
- Acyclovir/valacyclovir: Despite in vitro activity against EBV and reduction of oropharyngeal shedding, no clinical benefit in controlled trials for IM. When acyclovir is given, EBV clears from oropharynx but persists in B cells. Occasionally used in immunocompromised patients.
- HLH: Etoposide, cyclosporine, and/or corticosteroids.
- PTLD in transplant patients: Reduce immunosuppression; rituximab (anti-CD20) is used for treatment and prophylaxis.
2025 meta-analysis note: A recent systematic review (PMID 40512251, Eur J Clin Microbiol Infect Dis 2025) examined the rash associated with antibiotic administration in IM patients - confirming the well-known association.
- Harrison's 22E, p. 1555; Red Book 2021, p. 552
EBV-Associated Malignancies
Latent EBV infection is oncogenic when cellular immunity is inadequate:
| Malignancy | Notes |
|---|---|
| Burkitt lymphoma | >90% of endemic (African) cases; ~20% of sporadic cases; B-cell tumor with c-MYC translocation |
| Nasopharyngeal carcinoma | ~100% in endemic areas (Southeast Asia, Inuit); expresses EBNA1, LMP1 |
| Hodgkin lymphoma | EBV in Reed-Sternberg cells in up to 50% of cases |
| Post-transplant lymphoproliferative disorder (PTLD) | 1-10% of transplant recipients; highest risk in small intestine transplant |
| CNS lymphoma in AIDS | Virtually 100% EBV-associated |
| Gastric carcinoma | Subset of cases |
| T/NK-cell lymphomas | Less common |
| Multiple sclerosis | Large epidemiologic studies show strong association (mechanism unclear) |
- Harrison's 22E; Jawetz, Melnick & Adelberg's 28E; Red Book 2021
X-linked Lymphoproliferative Syndrome (XLP / Duncan disease)
A rare but important complication to know: Boys with mutations in SH2D1A (SAP protein) or XIAP/BIRC4 genes develop uncontrolled EBV infection, leading to fatal IM, HLH, or nodular B-cell lymphomas. A related condition, XMEN disease (MAGT1 gene mutation), is characterized by chronic high EBV DNAemia and heightened susceptibility to EBV-associated lymphomas.
- Red Book 2021, p. 548
Summary: Infectious mononucleosis is a clinical syndrome of EBV primary infection in adolescents/young adults, driven not by viral cytopathology alone but by the massive CD8+ T-cell response. Diagnosis rests on the clinical triad + Monospot or specific EBV serology. Management is supportive; splenic precautions and avoidance of amoxicillin are the two most actionable clinical pearls. The virus persists lifelong in memory B cells and, under immunosuppression, can drive lymphomagenesis.
Gilbert disease
Finding Sources
Finding Sources
Searching PubMed
"Gilbert syndrome" AND UGT1A1
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I have comprehensive data from multiple authoritative sources. Here is the full structured answer:
Gilbert Syndrome (Gilbert's Disease)
Definition
Gilbert syndrome is the most common hereditary cause of unconjugated hyperbilirubinemia - a benign, chronic, intermittent condition caused by impaired hepatic bilirubin conjugation. It carries an excellent prognosis and requires no treatment. Clinically, it is important mainly because it is frequently misidentified as liver disease.
- Tietz Textbook of Laboratory Medicine, 7th Ed; Harrison's 22E
Genetics and Molecular Basis
The defect lies in reduced activity of UDP-glucuronosyltransferase 1A1 (UGT1A1), the enzyme responsible for conjugating unconjugated (indirect) bilirubin with glucuronic acid in hepatocytes.
Mutation
- Most common (Western populations): A polymorphism in the 5' TATA box promoter region of the UGT1A1 gene - an insertion of an extra TA repeat, producing A(TA)7TAA instead of the normal A(TA)6TAA (the UGT1A1*28 allele). More TA repeats = less transcription factor binding = reduced UGT1A1 expression.
- The number of repeats is inversely proportional to transcriptional activity - patients with 8 repeats have higher bilirubin levels than those with 7.
- Asian populations: Point mutations in exon 1 (coding region) of UGT1A1 are more common than the *28 allele.
- Gene location: Chromosome 2.
- Result: UGT1A1 activity is reduced to approximately 10-35% of normal (roughly 30% retained activity is sufficient to prevent serious consequences).
- Inheritance: Autosomal recessive (homozygous *28/*28 genotype required for phenotypic expression).
Allele Frequencies of UGT1A1*28
| Population | Approximate allele frequency |
|---|---|
| Black/African | ~42% |
| White/European | ~30% |
| Asian | ~10% |
This explains why Gilbert syndrome is most prevalent in African populations and least common in Asians.
- Goldman-Cecil Medicine; Tietz Textbook of Laboratory Medicine; Yamada's Gastroenterology
Prevalence
- Affects approximately 3-10% of the general population (various sources report 3-7%, up to 6-12% in some studies).
- Males predominate over females by a ratio of 1.5-7:1 (estrogens may reduce bilirubin levels in females by altering metabolism, whereas androgens in adolescence increase bilirubin - which is why the condition often first presents in adolescence).
Pathophysiology
Normal bilirubin metabolism:
- Unconjugated bilirubin (from heme breakdown) is bound to albumin in plasma → taken up by hepatocytes → conjugated by UGT1A1 with glucuronic acid → water-soluble conjugated (direct) bilirubin → excreted into bile.
In Gilbert syndrome:
- UGT1A1 activity is reduced (~10-35% of normal).
- Unconjugated bilirubin accumulates in the blood.
- The conjugated bilirubin fraction remains normal → so urine bilirubin is absent (unconjugated bilirubin is not water-soluble and cannot be filtered by the kidney).
- Despite reduced total biliary bilirubin output, the ratio of bilirubin monoglucuronide to diglucuronide is increased, suggesting a secondary defect in converting mono- to diglucuronide also exists.
Clinical Features
-
Most patients are asymptomatic.
-
Diagnosis is often made incidentally on routine blood work.
-
Intermittent mild jaundice/scleral icterus may be the only sign, typically triggered by:
- Fasting or caloric restriction (most classic trigger - bilirubin rises 2-3 fold)
- Physiological stress
- Intercurrent illness/infection
- Physical exertion
- Alcohol use
- Dehydration
- Menstruation (in some females)
-
Symptoms that have been attributed to Gilbert syndrome (fatigue, abdominal discomfort) are generally not proven to be causally related.
-
No hepatomegaly, no splenomegaly, no stigmata of chronic liver disease.
-
Liver function tests are entirely normal except for elevated unconjugated bilirubin.
-
No bilirubin in urine (since unconjugated bilirubin is not renally excreted).
-
No anemia (unlike hemolysis, there is no increased bilirubin production).
-
Harrison's 22E, p. 1707; Tietz Textbook; Sleisenger & Fordtran's GI & Liver Disease
Laboratory Findings
| Test | Result in Gilbert Syndrome |
|---|---|
| Total serum bilirubin | Mildly elevated: typically 1.5-3 mg/dL (rarely up to 6 mg/dL) |
| Unconjugated (indirect) bilirubin | Elevated |
| Conjugated (direct) bilirubin | Normal |
| ALT, AST, ALP, GGT | Normal |
| Albumin, PT/INR | Normal |
| CBC | Normal (no hemolysis) |
| Urinary bilirubin | Absent |
| Urobilinogen (urine/stool) | Normal |
| Reticulocyte count | Normal |
Provocative tests (rarely needed now):
-
Fasting test: 400 kcal/day for 2 days → bilirubin rises by >50% above baseline; sensitivity ~90%. Rise seen in normal individuals too, but less pronounced.
-
Phenobarbital test: Phenobarbital induces UGT1A1 → bilirubin normalizes (used in both Gilbert's and Crigler-Najjar type II, but not type I).
-
Nicotinic acid (fasting) provocation: IV nicotinic acid causes a >3-fold rise in bilirubin in Gilbert's.
-
Tietz Textbook of Laboratory Medicine; Harrison's 22E
Differential Diagnosis of Isolated Unconjugated Hyperbilirubinemia
| Condition | Key Features |
|---|---|
| Gilbert syndrome | Benign; normal LFTs, no anemia; UGT1A1 promoter mutation; triggered by fasting |
| Hemolysis (autoimmune, hereditary spherocytosis, G6PD, etc.) | Anemia + reticulocytosis + elevated LDH + low haptoglobin |
| Crigler-Najjar type I | Neonatal severe jaundice >20 mg/dL; no UGT1A1 activity; kernicterus; fatal without LT |
| Crigler-Najjar type II | Bilirubin 6-25 mg/dL; responds to phenobarbital; usually survives to adulthood |
| Ineffective erythropoiesis | B12/folate/iron deficiency; thalassemia |
| Drug-induced | Rifampicin, probenecid reduce hepatic uptake |
Comparison of Hereditary Hyperbilirubinemias
| Feature | Gilbert | Crigler-Najjar I | Crigler-Najjar II | Dubin-Johnson | Rotor |
|---|---|---|---|---|---|
| Gene | UGT1A1 | UGT1A1 | UGT1A1 | MRP2 (ABCC2) | OATP1B1/1B3 |
| Defect | ↓ conjugation | No conjugation | ↓↓ conjugation | ↓ canalicular export | ↓ sinusoidal reuptake |
| Bilirubin type | Unconjugated | Unconjugated | Unconjugated | Mixed (conjugated) | Mixed (conjugated) |
| Bilirubin level | <3 mg/dL | >20 mg/dL | 6-25 mg/dL | <7 mg/dL | <7 mg/dL |
| Phenobarbital | Normalizes | No effect | Reduces | - | - |
| Liver histology | Normal | Normal | Normal | Coarse pigment | Normal |
| Prognosis | Normal | Death (infancy) | Usually normal | Normal | Normal |
- Sleisenger & Fordtran's GI & Liver Disease (Table)
Clinical Significance and Complications
While Gilbert syndrome is fundamentally benign, several important clinical interactions exist:
1. Drug Toxicity - Irinotecan (CPT-11)
- Most clinically important pharmacogenomic implication.
- Irinotecan's active metabolite SN-38 is detoxified by UGT1A1 glucuronidation to the inactive SN-38G.
- In Gilbert syndrome patients, reduced UGT1A1 activity → impaired SN-38 detoxification → accumulation of SN-38 → severe toxicity: intractable diarrhea, myelosuppression, neutropenia.
- Baseline unconjugated bilirubin level correlates with irinotecan/SN-38 AUC and neutropenia severity.
- UGT1A1 genotyping before irinotecan therapy is recommended (FDA label updated).
2. Raloxifene
- Also a UGT1A1 substrate; homozygous *28 allele results in ~2-fold higher drug exposure.
3. Acetaminophen
- Primarily metabolized by glucuronidation; theoretical increased susceptibility to hepatotoxicity in Gilbert syndrome (though evidence is not definitive).
4. HIV Protease Inhibitors
- Atazanavir and indinavir inhibit UGT1A1, causing a Gilbert-like hyperbilirubinemia. Patients with pre-existing Gilbert syndrome on these drugs may develop more pronounced jaundice.
5. Cardiovascular / Metabolic - Potential Protective Effects
- Bilirubin is an endogenous antioxidant - inhibits LDL oxidation.
- The Framingham Heart Study found that the UGT1A1*28 homozygous genotype (higher bilirubin) was associated with lower cardiovascular risk.
- Some studies also suggest reduced risk of diabetes and metabolic syndrome.
- Evidence is observational and not all studies agree.
6. Gallstones
- Increased unconjugated bilirubin load in bile may increase the risk of pigment gallstones.
7. Neonatal Jaundice
-
Neonates with UGT1A1 polymorphisms (especially homozygous *28) may have more pronounced and prolonged physiological jaundice, particularly if breastfed.
-
Goldman-Cecil Medicine; Goodman & Gilman's Pharmacology; Tietz Textbook; Sleisenger & Fordtran's
Diagnosis
Gilbert syndrome is a clinical diagnosis based on:
- Isolated mild unconjugated hyperbilirubinemia (bilirubin <6 mg/dL, almost entirely indirect)
- Normal LFTs (ALT, AST, ALP, GGT, albumin, PT)
- No hemolysis (normal CBC, reticulocyte count, LDH, haptoglobin)
- No urinary bilirubin
- Characteristic fluctuating course - worsens with fasting/illness
Genetic testing (UGT1A1 promoter analysis) can confirm but is not routinely required.
Liver biopsy is not indicated. (Histology is normal, or may show occasional lipofuscin pigment.)
Key distinguishing point from hemolysis: In hemolysis there is anemia + reticulocytosis + raised LDH + low haptoglobin. In Gilbert syndrome, all are normal.
Key distinguishing point from hepatitis: Gilbert syndrome has no urinary bilirubin, no elevation of ALT/AST, no systemic symptoms of liver disease.
Treatment
None required.
- Patients must be reassured that this is a benign condition, not a liver disease.
- No dietary restrictions are necessary in general (though very prolonged fasting can trigger jaundice).
- Phenobarbital can normalize bilirubin by inducing UGT1A1 but is not clinically indicated.
- Caution with irinotecan and other UGT1A1-metabolized drugs - dose adjustment or genotype-guided dosing should be considered.
- Standard precautions are appropriate if the patient is undergoing surgery (anesthesia teams should be aware since bilirubin levels may rise peri-operatively from fasting and stress).
Summary Table
| Feature | Gilbert Syndrome |
|---|---|
| Cause | UGT1A1 promoter mutation (A(TA)7TAA) |
| Enzyme defect | UGT1A1 reduced to ~10-35% of normal |
| Inheritance | Autosomal recessive |
| Prevalence | 3-10% of population; M > F |
| Bilirubin type | Unconjugated only |
| Bilirubin level | Usually <3 mg/dL; rises with fasting/stress |
| LFTs | Normal |
| Urine bilirubin | Absent |
| Liver biopsy | Normal |
| Treatment | None; reassurance |
| Prognosis | Excellent |
| Key drug interaction | Irinotecan toxicity (↑ SN-38 accumulation) |
Sources: Harrison's Principles of Internal Medicine 22E; Goldman-Cecil Medicine; Tietz Textbook of Laboratory Medicine 7E; Sleisenger & Fordtran's GI & Liver Disease; Yamada's Textbook of Gastroenterology; Harper's Illustrated Biochemistry 32E; Goodman & Gilman's Pharmacological Basis of Therapeutics.
This is a shared conversation. Sign in to Orris to start your own chat.