I have my university exam of 1 st year of subject biochemistry These are the important long answer give me answer of this question"Galactose metabolism" and remeber it's a university examination
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed for a thorough university-level answer. Let me compose it.
Galactose Metabolism
1. Introduction
Galactose is a six-carbon aldohexose (C-4 epimer of glucose) that enters the body primarily as lactose (galactosyl β-1,4-glucose) from milk and dairy products. Intestinal lactase (β-galactosidase) hydrolyses lactose into free glucose and free galactose, which are then absorbed by the small intestinal epithelium. A smaller amount of galactose is also released during lysosomal degradation of glycoproteins and glycolipids. Unlike glucose, galactose transport into cells is not insulin-dependent.
The liver is the main site of galactose catabolism, where the sugar is converted to glucose 1-phosphate and fed into mainstream glucose metabolism at the cost of only one ATP.
2. Sources of Galactose
| Source | Details |
|---|---|
| Dietary (exogenous) | Lactose in milk/dairy, hydrolysed by lactase → glucose + galactose |
| Endogenous | Lysosomal breakdown of glycoproteins, glycolipids, glycosaminoglycans |
3. The Leloir Pathway (Main Catabolic Pathway)
The Leloir pathway converts galactose to glucose 1-phosphate in three enzyme-catalysed steps.
Step 1 - Phosphorylation by Galactokinase
Galactose + ATP → Galactose 1-phosphate + ADP
- Enzyme: Galactokinase (GALK)
- Location: Most tissues, especially liver
- This is the committed, ATP-dependent first step
- Galactose 1-phosphate cannot enter glycolysis directly - it must be activated further
Step 2 - Exchange Reaction (Uridylyltransferase)
Galactose 1-phosphate + UDP-glucose → UDP-galactose + Glucose 1-phosphate
- Enzyme: Galactose 1-phosphate uridylyltransferase (GALT, also written GALT or Gal-1-PUT)
- The phosphate oxygen on galactose 1-phosphate attacks the α-phosphate of UDP-glucose
- This is an exchange (dismutation) reaction - no energy is consumed or released in this step
- Glucose 1-phosphate produced is isomerized to glucose 6-phosphate by phosphoglucomutase, then enters glycolysis or is released as free glucose by the liver
Step 3 - Epimerization by UDP-hexose 4-Epimerase
UDP-galactose → UDP-glucose
- Enzyme: UDP-glucose 4-epimerase (also called UDP-hexose 4-epimerase or GALE)
- Reverses the configuration of the hydroxyl group at C-4
- This regenerates UDP-glucose for use in the uridylyltransferase reaction (Step 2), making the pathway catalytic with respect to UDP-glucose
- The reaction is reversible - this is also how the body synthesizes UDP-galactose from glucose when dietary galactose is absent
Net equation:
Galactose + ATP → Glucose 1-phosphate + ADP
Pathway Diagram
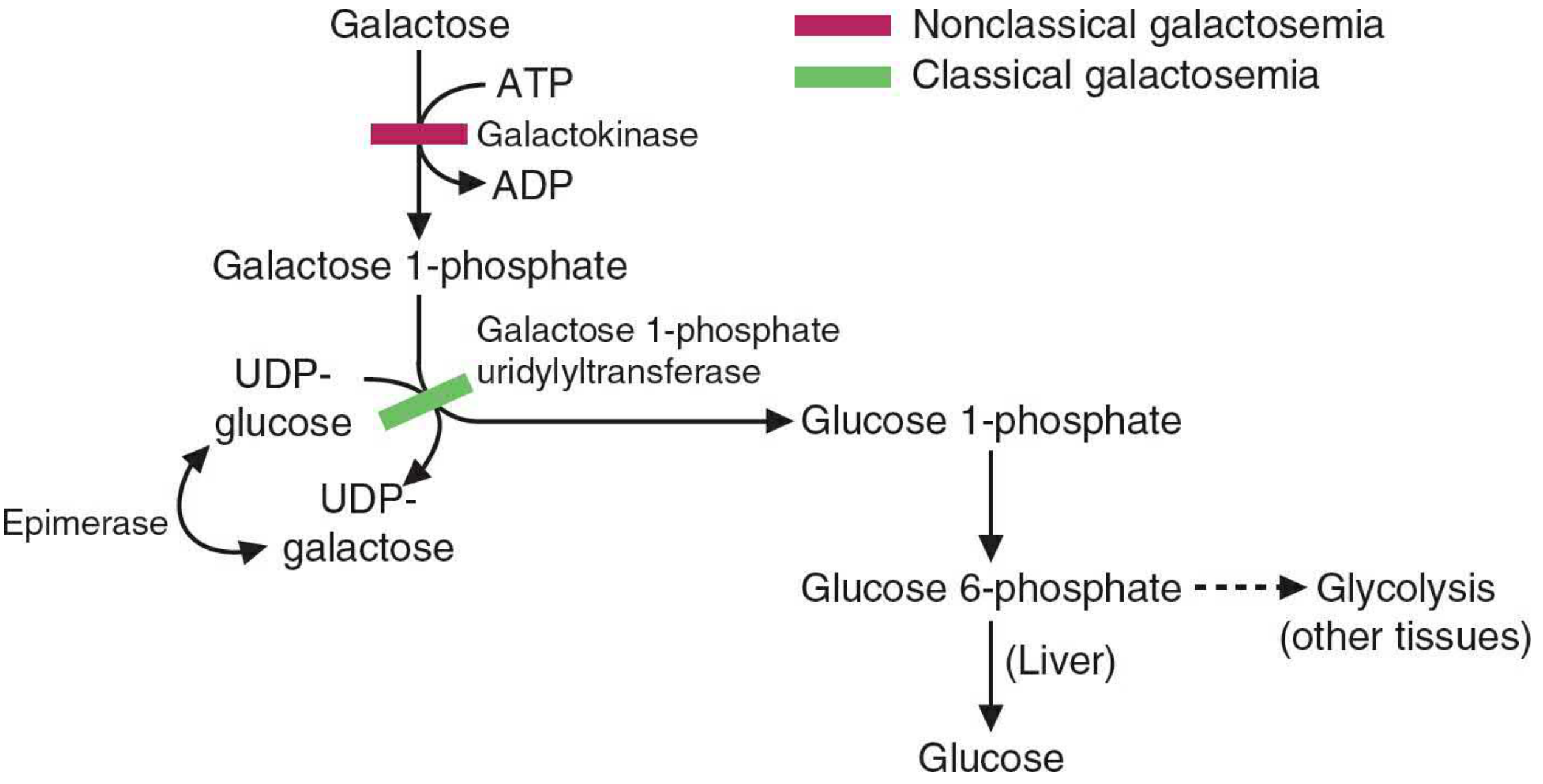
Pink bar = enzyme deficient in nonclassical (Type II) galactosemia; Green bar = enzyme deficient in classical (Type I) galactosemia
4. Fate of Products
| Product | Fate |
|---|---|
| Glucose 1-phosphate | Isomerized to glucose 6-phosphate → glycolysis, glycogenesis, or (in liver) free glucose |
| UDP-galactose | Used in biosynthetic reactions (see below) OR epimerized back to UDP-glucose |
5. Biosynthetic Role of UDP-Galactose
UDP-galactose is not merely a metabolic intermediate - it is an activated sugar nucleotide that serves as a galactose donor in several important anabolic processes:
- Lactose synthesis (mammary glands): UDP-galactose + glucose → lactose (catalysed by lactose synthase, which requires α-lactalbumin as regulatory B-subunit)
- Glycoprotein synthesis: Galactose residues are added to N-linked and O-linked oligosaccharide chains
- Glycolipid synthesis: Galactosylceramide and other glycolipids (important in myelin)
- Glycosaminoglycan synthesis: e.g., keratan sulphate, chondroitin sulphate
Note: Even when dietary galactose is absent, UDP-galactose can be synthesized de novo by the reverse action of UDP-glucose 4-epimerase acting on UDP-glucose derived from glycolysis. This ensures all biosynthetic needs are met.
6. Alternate Pathway - Galactitol Formation (Polyol/Sorbitol Pathway)
When galactose accumulates in excess (e.g., in enzyme deficiency states), it is reduced to galactitol by aldose reductase (same enzyme that reduces glucose to sorbitol):
Galactose + NADPH → Galactitol + NADP⁺
- Galactitol cannot be further metabolized and accumulates in the lens, liver, kidney, and brain
- Accumulation of galactitol in the lens osmotically draws water in → cataract formation
- This pathway is the major cause of tissue damage in galactosemia
7. Disorders of Galactose Metabolism (Galactosemia)
Galactosemia is an autosomal recessive inborn error of carbohydrate metabolism. There are four recognized types:
Type I - Classical Galactosemia (Most Severe)
- Deficient enzyme: Galactose 1-phosphate uridylyltransferase (GALT)
- Accumulates: Galactose 1-phosphate AND galactose → galactitol
- Clinical features:
- Neonatal period: vomiting, jaundice (conjugated hyperbilirubinemia), hepatomegaly, sepsis (especially E. coli neonatal sepsis), failure to thrive
- Later: cataracts, aminoaciduria, cirrhosis, intellectual disability
- Females: premature ovarian insufficiency
- Even with early treatment: developmental delays, speech problems, and motor dysfunction can persist
- Treatment: Eliminate galactose and lactose from diet (galactose-free formula); the body can still synthesize UDP-galactose via the epimerase reverse reaction
Type II - Galactokinase Deficiency (Nonclassical)
- Deficient enzyme: Galactokinase (GALK)
- Accumulates: Galactose → galactitol (galactose 1-phosphate does NOT accumulate)
- Clinical features: Primarily cataracts (bilateral); pseudotumor cerebri in some cases
- Milder than Type I because galactose 1-phosphate does not accumulate
- Treatment: Lactose-free diet; cataracts may regress
Type III - Epimerase Deficiency (GALE Deficiency)
- Deficient enzyme: UDP-galactose 4-epimerase
- Two forms:
- Benign (limited to red/white blood cells): asymptomatic
- Severe (generalized): similar to classical galactosemia + hypotonia + sensorineural hearing loss
- Accumulates both galactose 1-phosphate and UDP-galactose in severe form
Type IV - Galactose Mutarotase Deficiency (GALM)
- Deficient enzyme: Galactose mutarotase (converts β-galactose to α-galactose, which is the substrate for galactokinase)
- Clinical features: Cataracts
- Identified primarily in Japan; considered mild
Summary Table of Galactosemia Types
| Type | Enzyme Deficient | Accumulated Metabolites | Key Clinical Features |
|---|---|---|---|
| I (Classical) | GALT (uridylyltransferase) | Galactose 1-P, galactose, galactitol | Jaundice, liver failure, cataracts, sepsis, intellectual disability |
| II | GALK (galactokinase) | Galactose, galactitol | Cataracts (mainly) |
| III | GALE (epimerase) | UDP-galactose, galactose 1-P | Varied: mild to severe like Type I |
| IV | GALM (mutarotase) | Galactose | Cataracts (mild) |
8. Diagnosis
- Newborn screening: Reduced GALT activity (or elevated galactose/galactose 1-phosphate) on heel-prick blood spot - classical galactosemia is part of newborn screening panels
- Urine: Reducing substances positive (galactose is a reducing sugar); but negative with glucose-specific dipstick
- Enzyme assay: Erythrocyte GALT, GALK, or GALE activity
- Genetic testing: Mutation analysis of GALT, GALK, GALE genes
9. Clinical Connection - Neonatal E. coli Sepsis
A classical, high-yield clinical association: neonates with undiagnosed galactosemia are at markedly increased risk for gram-negative sepsis (especially E. coli). The exact mechanism is not fully established but may relate to impaired neutrophil function due to galactose 1-phosphate accumulation in leukocytes.
10. Summary
The key points to remember for your exam:
- Dietary source: Lactose (milk) → glucose + galactose by lactase
- Three-step Leloir pathway: Galactokinase → Uridylyltransferase → Epimerase → produces glucose 1-phosphate
- Net cost: 1 ATP per galactose molecule
- UDP-galactose is the activated form used for biosynthesis (lactose, glycoproteins, glycolipids)
- Overflow pathway: Excess galactose → galactitol (by aldose reductase) → accumulates → cataracts
- Type I galactosemia (GALT deficiency) is the most severe; Type II (GALK deficiency) causes mainly cataracts
- Treatment: Lifelong dietary elimination of galactose/lactose
Sources: Lippincott Illustrated Reviews: Biochemistry, 8th ed.; Basic Medical Biochemistry: A Clinical Approach, 6th ed.; Harrison's Principles of Internal Medicine, 22nd ed.
This is a shared conversation. Sign in to Orris to start your own chat.