Epileptiform fits
Reading File
Checking Date & Time
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Searching PubMed
epilepsy seizure management guidelines
Here is a comprehensive overview of epileptiform fits (seizures and epilepsy):
Epileptiform Fits (Seizures & Epilepsy)
Definition
A seizure is a transient occurrence of signs or symptoms due to abnormal, excessive or synchronous neuronal activity in the brain. The term "epileptiform fit" encompasses this concept — the word epilepsy derives from the Greek meaning "to seize upon."
Epilepsy is the condition of recurrent unprovoked seizures due to a chronic underlying process. By definition, this requires ≥2 unprovoked seizures (though a single seizure with high-risk EEG/imaging features may establish the diagnosis). The worldwide prevalence is 5–30 per 1000 persons.
A single seizure or brief burst during a medical illness (e.g., fever, electrolyte disturbance) does not necessarily constitute epilepsy. — Harrison's Principles of Internal Medicine 22E
Classification (ILAE 2017)
| Onset Type | Subtypes |
|---|---|
| Focal onset | Intact awareness / Impaired awareness; Motor or nonmotor onset; May evolve to bilateral tonic-clonic |
| Generalized onset | Motor (tonic-clonic, atonic, myoclonic); Nonmotor (absence) |
| Unknown onset | Motor, nonmotor, or unclassified |
(Note: "Partial seizures" and "simple/complex partial" terminology has been retired.)
Types of Seizures
1. Focal Onset Seizures
Arise from a discrete network in one hemisphere.
- With intact awareness: The patient remains conscious. Motor forms include tonic, clonic, or myoclonic movements contralateral to the focus (e.g., involuntary left hand movements from a right motor cortex focus). Sensory forms include numbness/tingling (parietal lobe), olfactory hallucinations ("uncinate fits" — unpleasant smells from medial temporal lobe), visual flashes (occipital lobe), or auditory phenomena (superior temporal gyrus).
- With impaired awareness (formerly complex partial): Typically temporal lobe origin. The patient stares blankly and performs automatisms (lip smacking, picking movements). There may be a prior aura (e.g., rising epigastric sensation, déjà vu, fear).
- Focal to bilateral tonic-clonic: A focal seizure that evolves into full generalized convulsion — previously called "secondary generalisation."
2. Generalized Onset Seizures
Tonic-clonic (grand mal): The classic convulsion:
- Tonic phase (10–20 s): Sudden loss of consciousness, fall, brief flexion then sustained extension of the body, forced air expulsion ("epileptic cry"), cyanosis, pupils dilated and fixed
- Clonic phase (~30 s): Rhythmic jerking at ~8/s, coarsening to 4/s, then subsiding; autonomic features prominent (tachycardia, hypertension, salivation, incontinence)
- Postictal phase: Deep stupor then gradual confusion, headache, myalgia, amnesia for the event
Absence (petit mal): Brief (~10 s) blank stare with sudden onset and offset, no postictal state. EEG shows classic 3-Hz spike-and-wave discharges. Common in childhood, often precipitated by hyperventilation.
Atonic seizures ("drop attacks"): Sudden loss of muscle tone, causing falls.
Myoclonic seizures: Brief, shock-like involuntary muscle contractions. Characteristic of juvenile myoclonic epilepsy (JME).
Infantile spasms (West syndrome): Gross flexion/extension spasms in infants <1 year; EEG shows hypsarrhythmia (chaotic high-amplitude spike-wave). Treated with ACTH or vigabatrin.
Pathophysiology
The fundamental mechanism is a disruption of the balance between excitation (glutamate) and inhibition (GABA):
- Ictal discharge initiation: Involves changes in ion channel conductance, receptor sensitivity, and extracellular ion concentrations. A paroxysmal depolarisation shift (PDS) in individual neurons, with synchronous firing in a population of neurons.
- Absence seizures: Linked to oscillatory thalamocortical circuits involving GABA_A receptors, T-type Ca²⁺ channels, and K⁺ channels. Genetic mutations in these components are well established.
- Epileptogenesis: The process by which a normal neural network becomes chronically hyperexcitable, often after an initial CNS injury (trauma, stroke, infection) with a delay of months to years before the first seizure. Structural changes include hippocampal neuronal loss and mossy fibre sprouting.
- Genetic channelopathies: Many idiopathic epilepsies are now known to involve mutations in ion channel genes — notably SCN1A (Dravet syndrome), KCNQ2, CHRNA4, and GABA receptor subunits.
Diagnosis
EEG
The key diagnostic tool. Epileptiform activity consists of spikes or sharp waves, which may appear interictally or ictally:
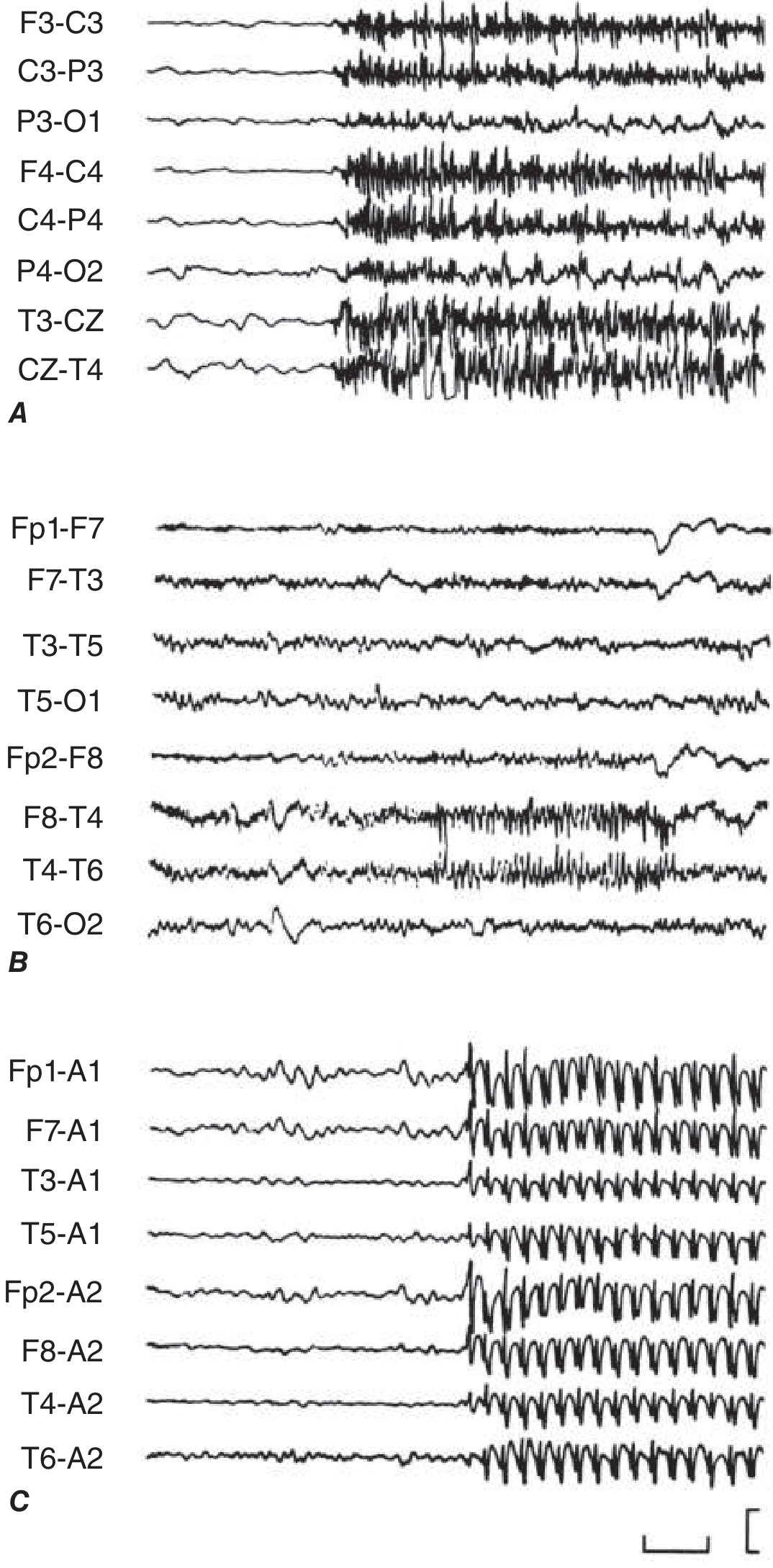
Figure: Electrographic seizures. A. Generalised tonic seizure showing synchronous sharp activity. B. Right temporal focal discharge during impaired awareness. C. 3-Hz spike-wave of absence seizure. — Harrison's 22E
- Normal interictal EEG does not exclude epilepsy
- Epileptiform spikes/sharp waves predict higher recurrence risk
- 3-Hz spike-wave: Absence epilepsy
- Video-EEG monitoring is the gold standard for seizure characterisation
- High-density EEG (up to 257 electrodes) and MEG allow source localisation for surgical planning
Neuroimaging
- MRI is preferred — detects hippocampal sclerosis, cortical dysplasia, tumours, vascular malformations
- PET/SPECT for presurgical evaluation of refractory epilepsy
Blood tests
Glucose, electrolytes (Na⁺, Ca²⁺, Mg²⁺), renal/liver function, toxicology screen — to identify reversible/provoked causes.
Aetiology
| Category | Examples |
|---|---|
| Genetic/Idiopathic | JME, childhood absence, Dravet syndrome, benign rolandic epilepsy |
| Structural | Cortical dysplasia, hippocampal sclerosis, tumour, vascular malformation, stroke, trauma |
| Metabolic | Hypoglycaemia, hyponatraemia, hypocalcaemia, uraemia |
| Infective | Meningitis, encephalitis, cerebral abscess, neurocysticercosis |
| Immune | Anti-NMDA receptor encephalitis, LGI1 antibody encephalitis |
| Provoked (acute symptomatic) | Alcohol withdrawal, drug toxicity, eclampsia, hypoxia |
Special Situations
Status Epilepticus
Seizure activity lasting >5 minutes (convulsive) or >10–15 minutes (absence/focal), or recurrent seizures without recovery between. A neurological emergency with significant mortality.
Management:
- 0–5 min: ABC, IV access, glucose check, thiamine if nutritionally at risk
- 5–20 min: IV/IM benzodiazepine (lorazepam 0.1 mg/kg IV; or midazolam 10 mg IM)
- 20–40 min: IV levetiracetam, valproate, or fosphenytoin
- >40 min (refractory): Anaesthetic agents (propofol, midazolam, thiopental) with ICU intubation
Febrile Seizures
- Simple: Single generalised tonic-clonic, <15 min, in child 6 months–5 years with fever; benign prognosis
- Complex: Focal, prolonged (>15 min), or recurrent within 24 h; higher risk of future epilepsy
- Temperature usually >38°C; herpesvirus 6 is a common precipitant
Treatment: Antiseizure Drug (ASD) Selection
| Seizure Type | First-Line Drugs |
|---|---|
| Generalised tonic-clonic | Lamotrigine, valproic acid, levetiracetam |
| Focal | Lamotrigine, carbamazepine, levetiracetam, oxcarbazepine |
| Typical absence | Valproic acid, ethosuximide, lamotrigine |
| Atypical absence, myoclonic, atonic | Valproic acid, lamotrigine, levetiracetam |
Key principles:
- Start with monotherapy; if inadequate at maximum tolerated dose, switch to another monotherapy before adding a second agent
- Initiate treatment after a second unprovoked seizure, or after a first seizure with high recurrence risk (structural lesion, abnormal EEG, nocturnal onset)
- Common dose-related side effects: sedation, ataxia, dizziness, diplopia
- Idiosyncratic risks: rash (carbamazepine, lamotrigine), hepatotoxicity (valproate), bone marrow suppression
- Valproate: teratogenic — avoid in women of childbearing age unless no alternative
- Drug-resistant epilepsy (failure of ≥2 appropriate ASDs): refer for surgical evaluation; other options include responsive neurostimulation (RNS), vagus nerve stimulation (VNS), ketogenic diet
When to Stop Driving / Lifestyle Advice
Patients should be counselled on:
- Driving restrictions (typically seizure-free for 6–12 months depending on jurisdiction)
- Avoiding unsupervised swimming, bathing, heights, and operating heavy machinery
- Sleep deprivation and alcohol as seizure triggers
- Contraception/pregnancy planning (enzyme-inducing ASDs affect oral contraceptives; folate supplementation mandatory)
Sources:
- Harrison's Principles of Internal Medicine 22E, Chapter 436 — Seizures and Epilepsy
- Adams and Victor's Principles of Neurology 12E — Epilepsy and Other Seizure Disorders
- Recent systematic review on status epilepticus management guidelines (Vignatelli et al., Epilepsia 2024, PMID: 38606469)
This is a shared conversation. Sign in to Orris to start your own chat.