Vitamin B12
Reading File
Finding Sources
Finding Sources
Searching PubMed
"vitamin B12" AND deficiency
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have rich content from multiple authoritative textbooks. Let me compile the comprehensive answer.
Vitamin B12 (Cobalamin)
Structure and Chemistry
Vitamin B12 is a water-soluble vitamin with a cobalt atom at the center of a corrin ring - its defining feature. It exists in several chemical forms:
- Adenosylcobalamin (adocobalamin) - the main form in mitochondria; serves as cofactor for L-methylmalonyl-CoA mutase
- Methylcobalamin - the form in human plasma and cell cytoplasm; cofactor for methionine synthase
- Hydroxocobalamin - a minor form that is rapidly converted to the above two by exposure to light
- Cyanocobalamin - the synthetic pharmaceutical form
(Harrison's Principles of Internal Medicine, 22nd ed., p. 824)
Dietary Sources and Requirements
Cobalamin is synthesized exclusively by microorganisms - no plant or animal tissue can make it. The only dietary sources for humans are animal-origin foods: meat, fish, eggs, and dairy products. Vegetables and fruits contain essentially no cobalamin unless bacterially contaminated.
| Parameter | Value |
|---|---|
| Normal Western diet intake | 5-30 µg/day |
| Daily losses (urine + feces) | 1-3 µg (~0.1% of stores) |
| Daily requirement | ~1-3 µg |
| US RDA (adults) | 2.4 µg/day |
| Pregnancy | 2.6 µg/day |
| Lactation | 2.8 µg/day |
| Total body stores | ~2-3 mg (sufficient for 3-4 years if intake stops) |
Because of efficient enterohepatic circulation (0.5-5 µg recycled through bile daily), a vegan will take years to become deficient, whereas someone with malabsorption (who cannot reabsorb biliary cobalamin) will develop deficiency much faster.
(Harrison's, p. 824; Goldman-Cecil Medicine)
Absorption - Step by Step
Cobalamin absorption is a highly orchestrated multistep process:
-
Gastric phase: Pepsin and gastric acid release cobalamin from food proteins. It immediately binds to haptocorrin (HC) / R-protein from saliva.
-
Duodenal phase: Pancreatic trypsin digests the HC-cobalamin complex. Cobalamin is released and transfers to intrinsic factor (IF), secreted by gastric parietal cells. IF is specific for true cobalamin and does not bind analogues.
-
Ileal phase: The IF-cobalamin complex binds the cubam receptor (cubilin + amnionless) on ileal enterocyte microvilli. The complex is internalized by endocytosis via clathrin-coated pits.
-
Intracellular export: Two exporters, products of the LMBD1 and ABCD4 genes, are needed to export cobalamin from lysosomes into the cytoplasm.
-
Portal delivery: After ~6 hours, cobalamin appears in portal blood bound to transcobalamin II (TC II), the physiologic delivery protein.
A second (passive) mechanism exists via buccal, duodenal, and ileal mucosa, but is extremely inefficient (<1% absorbed) - this is the basis for high-dose oral therapy in deficiency.
(Harrison's, p. 824; Goldman-Cecil, p. 1729)
Transport in Blood
| Carrier | Key Features |
|---|---|
| Haptocorrin / TC I | Carries ~80% of blood cobalamin; binds tightly; binds cobalamin analogues; removed by liver asialoglycoprotein receptors; derived from neutrophil granules; not the functional delivery protein |
| Transcobalamin II (TC II) | Carries only 10-20% of circulating cobalamin; the true delivery protein; synthesized by liver, macrophages, ileum, endothelium; delivers cobalamin to marrow, placenta, and tissues via TC II receptor / megalin (LRP-2) |
This is clinically important: total serum B12 mainly reflects TC I-bound cobalamin; TC II-bound ("holotranscobalamin") is the biologically active fraction.
(Harrison's, p. 824; Goldman-Cecil, p. 1730)
Biochemical Functions
Only two reactions in humans require cobalamin:
1. Methionine Synthesis (cytoplasm)
- Enzyme: Methionine synthase
- Cofactor: Methylcobalamin
- Reaction: Homocysteine + 5-methyltetrahydrofolate (5-MTHF) → Methionine + THF
- This regenerates THF, which is needed for purine and pyrimidine synthesis (DNA replication)
- This is the biochemical link between cobalamin and folate: cobalamin deficiency "traps" folate as 5-MTHF (the "methylfolate trap"), causing functional folate deficiency even when dietary folate is normal
2. Methylmalonyl-CoA Isomerization (mitochondria)
- Enzyme: Methylmalonyl-CoA mutase
- Cofactor: Adenosylcobalamin
- Reaction: L-methylmalonyl-CoA → Succinyl-CoA (part of propionate metabolism and odd-chain fatty acid oxidation)
- Deficiency causes methylmalonic acid (MMA) accumulation - a key diagnostic marker
(Harrison's, p. 831; Guyton & Hall Medical Physiology)
Causes of Deficiency
Dietary Insufficiency
- Vegans / strict vegetarians (no animal products)
- Breast-fed infants of deficient mothers
- Poverty-related dietary restriction (30-50% prevalence in parts of South America, Africa, India)
Malabsorption (most common cause of severe deficiency)
| Cause | Mechanism |
|---|---|
| Pernicious anemia | Autoimmune destruction of gastric parietal cells → loss of IF secretion; immune attack targets gastric H/K-ATPase |
| Gastric surgery / gastrectomy | Loss of parietal cell mass; loss of IF |
| Atrophic gastritis | Common in elderly (up to 20% of older adults); protein-bound cobalamin malabsorption |
| Chronic pancreatitis | Failure to digest HC → cobalamin not transferred to IF |
| Ileal disease / resection | Loss of cubilin/cubam receptor absorption site (Crohn's disease, surgical resection) |
| Metformin use | Reduces uptake via cubam receptor (calcium-dependent mechanism) |
| Nitrous oxide (N₂O) | Irreversibly oxidizes methylcobalamin; acute functional deficiency |
| Helicobacter pylori infection | Contributes to gastric atrophy |
Pernicious anemia incidence: 1-50 per 100,000/year; prevalence 50-4,000/100,000; more common in African and European ancestry; increases with age.
(Harrison's; Goldman-Cecil)
Clinical Features of Deficiency
Hematologic
- Megaloblastic anemia: Oval macrocytes, anisocytosis, poikilocytosis; MCV typically >100 fL
- Hypersegmented neutrophils (>5 nuclear lobes) - pathognomonic
- Leukopenia (usually >1.5 × 10⁹/L)
- Thrombocytopenia (occasionally)
- Bone marrow: hypercellular with abnormally large, immature erythroblasts ("megaloblasts") - nuclear-cytoplasmic maturation asynchrony
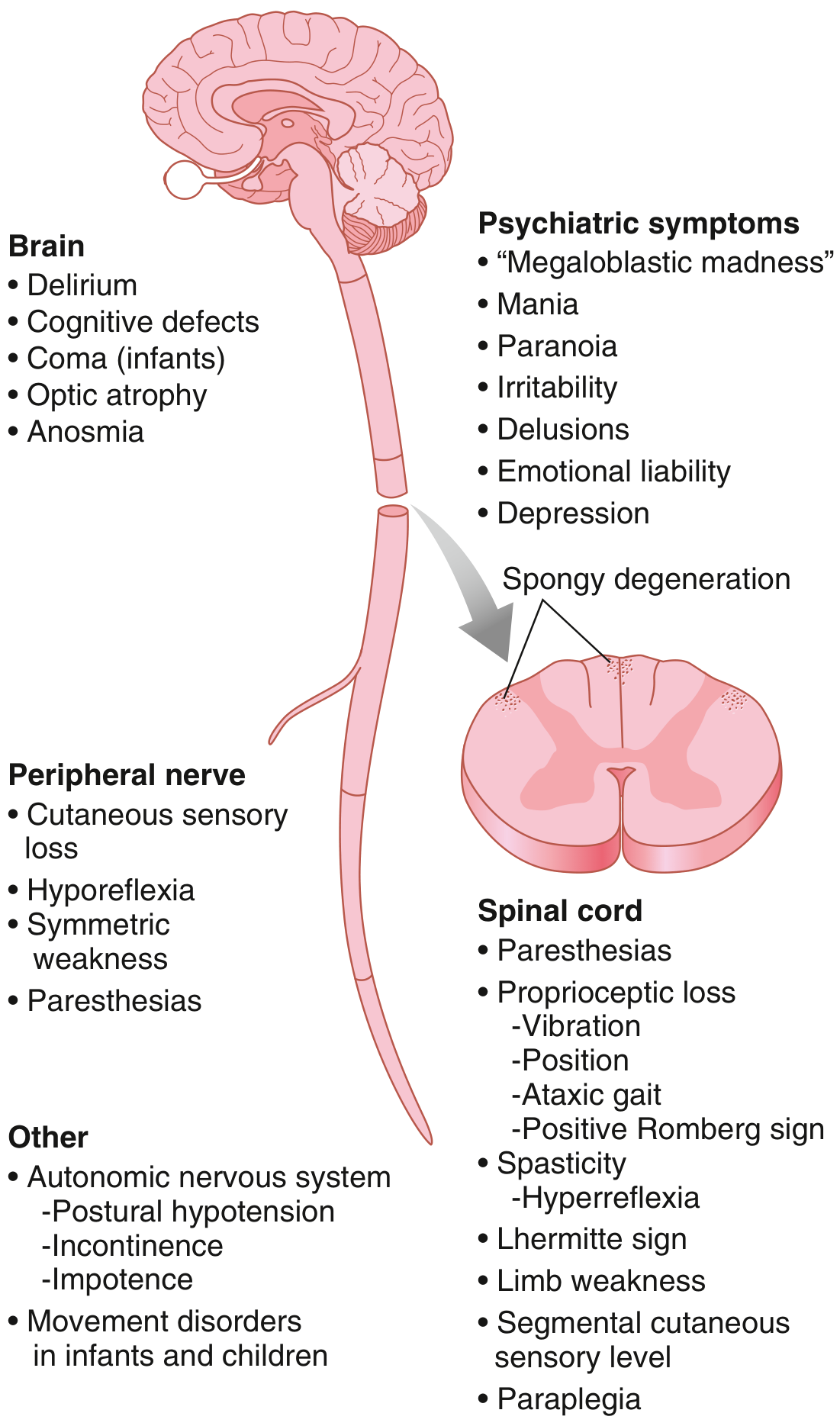
Neurological - Subacute Combined Degeneration of the Cord
This is the most distinctive neurological complication of B12 deficiency, involving demyelination of:
- Posterior columns (dorsal columns): loss of vibration sense and proprioception, sensory ataxia
- Lateral columns (corticospinal tracts): upper motor neuron signs, spasticity
- Peripheral neuropathy: symmetrical sensory loss, paresthesias
- In severe cases: paraplegia, cognitive impairment
Importantly, neurological damage can occur without anemia, especially when MCV is masked by co-existing iron deficiency.
Other Features
- Glossitis (smooth, red, painful tongue - "Hunter's glossitis")
- Angular cheilitis
- Elevated homocysteine → associated with cardiovascular risk
- Hyperpigmentation (in dark-skinned individuals)
- Jaundice (ineffective erythropoiesis with intramedullary hemolysis)
(Guyton & Hall; Harrison's; Goldman-Cecil)
Diagnosis
| Test | Details |
|---|---|
| Serum cobalamin | Normal: 118-738 pmol/L (160-1000 ng/L); <74 pmol/L (<100 ng/L) in deficiency; 74-148 pmol/L borderline. Note: anti-IF antibodies in pernicious anemia can cause false-normal levels in up to 50% of cases |
| Serum methylmalonic acid (MMA) | Elevated in cobalamin deficiency even before hematologic changes; falsely elevated in renal failure |
| Serum homocysteine | Elevated in both B12 and folate deficiency; less specific |
| Holotranscobalamin (active B12) | TC II-bound fraction; emerging as more specific marker |
| Anti-intrinsic factor antibodies | Highly specific for pernicious anemia |
| Anti-parietal cell antibodies | Sensitive but not specific for PA |
(Harrison's, pp. 832-833)
Treatment
Pernicious Anemia / Malabsorption
- Intramuscular cyanocobalamin or hydroxocobalamin: 1,000 µg daily for 1 week, then weekly for 1 month, then monthly (lifelong in PA)
- Hydroxocobalamin is preferred over cyanocobalamin in many countries because it is retained longer in the body
- High-dose oral cobalamin (1,000-2,000 µg/day) is effective even without IF, because ~1% is absorbed passively - now an accepted alternative in patients who prefer oral therapy
Food Malabsorption (elderly atrophic gastritis)
- Low-dose oral cyanocobalamin 50 µg/day may be sufficient
Dietary Deficiency
- Dietary modification or oral supplementation
Monitoring Response
- Reticulocytosis peaks at days 5-7 after starting treatment
- Neurological recovery depends on duration and severity - may be incomplete if treatment is delayed
Critical warning: Never give folate alone without first excluding B12 deficiency. Folate can correct the anemia but will allow neurological damage to progress ("masking").
(Harrison's, p. 832; Goldman-Cecil)
Key Drug Interactions
| Drug | Effect on B12 |
|---|---|
| Metformin | Reduces ileal absorption via cubam receptor |
| Proton pump inhibitors / H2 blockers | Reduce gastric acid → impair release from food protein |
| Nitrous oxide | Irreversible oxidation of cobalamin; acute depletion |
| Colchicine | Impairs ileal uptake |
Summary of Diagnostic Pathway
Suspected B12 deficiency
↓
Serum cobalamin + MMA + homocysteine
↓
Low B12 / high MMA → Confirmed deficiency
↓
Determine cause: dietary vs. malabsorption
↓
Anti-IF antibody + anti-parietal cell Ab
↓
Positive → Pernicious anemia → lifelong IM/oral replacement
Negative → Consider gastric/ileal disease, drugs
Sources:
- Harrison's Principles of Internal Medicine, 22nd Edition (2025), pp. 824-833
- Goldman-Cecil Medicine International Edition, pp. 1729-1730
- Guyton and Hall Textbook of Medical Physiology
- Recent systematic review: B12 status in vegans (Niklewicz et al., Nutr Bull, Dec 2024 - PMID 39373282) confirms substantially lower functional B12 status in adult vegans
Compare pernicious anemia vs. dietary B12 deficiency: clinical differences
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Here is the detailed comparison, drawn from multiple authoritative sources.
Pernicious Anemia vs. Dietary B12 Deficiency: Clinical Comparison
The Core Distinction
Both conditions cause B12 deficiency, but through entirely different mechanisms, and this shapes virtually every clinical difference between them.
| Pernicious Anemia (PA) | Dietary B12 Deficiency | |
|---|---|---|
| Root cause | Autoimmune destruction of gastric parietal cells → loss of intrinsic factor (IF) | Insufficient intake of animal-source foods |
| Mechanism of deficiency | Malabsorption - cannot absorb B12 regardless of how much is eaten | Inadequate supply to an otherwise intact GI tract |
| Enterohepatic cycle intact? | No - biliary B12 is also lost (5-9 µg/day lost vs. normal ~1 µg/day) | Yes - biliary B12 is efficiently reabsorbed |
Speed and Severity of Onset
This is one of the most clinically significant differences.
Pernicious anemia progresses faster and more severely because:
- Biliary cobalamin (0.5-5 µg/day) cannot be reabsorbed - there is no IF in the duodenum to bind it
- So instead of losing ~1 µg/day, PA patients lose 5-9 µg/day, dramatically shortening the time to depletion
- Body stores of 2-3 mg can be depleted in months to a few years
Dietary deficiency progresses slowly, taking years to decades:
- Enterohepatic circulation remains intact - biliary B12 keeps recycling
- A vegan will take years to exhaust stores even with zero dietary intake
- Deficiency often detected incidentally or with only subtle symptoms
(Goldman-Cecil Medicine, p. 1730; Harrison's, p. 824)
Demographics and Risk Groups
| Feature | Pernicious Anemia | Dietary Deficiency |
|---|---|---|
| Typical age | Middle-aged to elderly (mean 59-62 years; 10% in their 30s-40s) | Any age; depends on diet |
| Who gets it | Previously thought Northern European; now seen across all ethnicities | Vegans, strict vegetarians, breastfed infants of vegan mothers, populations in South Asia/Africa/South America with low meat intake |
| Sex | Slight female predominance | No sex predilection |
| Prevalence | ~1-2% of population; ~30% of all B12 deficiency cases | 30-50% prevalence in parts of South America, Africa, India |
| Family history | Significant - first-degree relatives have ~30% rate of anti-parietal cell antibodies | Not heritable; linked to cultural/socioeconomic factors |
Associated Conditions
This is a key distinguishing feature unique to PA:
Pernicious anemia is an autoimmune disease and clusters with other autoimmune conditions:
- Autoimmune thyroid disease (Graves' disease and Hashimoto's) - ~25% have both
- Type 1 diabetes mellitus
- Addison's disease (adrenal insufficiency)
- Vitiligo
- HLA associations: DRB1*03 and DRB1*04 genotypes
Also unique to PA: elevated gastric cancer risk - relative risk of gastric adenocarcinoma is 6.8 (95% CI 2.6-18.1). Up to 10% of PA patients develop gastric carcinoid tumors. Endoscopy is recommended at least once for screening.
Dietary deficiency has no such autoimmune or oncologic associations. Co-deficiencies of iron, folate, zinc, and other nutrients are common, especially in resource-limited settings.
(Goldman-Cecil, p. 1730; Henry's Clinical Diagnosis, p. 1244)
Gastric Features (Unique to PA)
| Finding | PA | Dietary Deficiency |
|---|---|---|
| Gastric parietal cell antibodies | 80-90% early; 55% advanced | Absent |
| Anti-intrinsic factor antibodies (Type I blocking, Type II binding) | 50-70%; highly specific | Absent |
| Achlorhydria (histamine-fast) | Present; very specific for PA | Absent |
| Serum gastrin | Markedly elevated (compensatory) | Normal |
| Pepsinogen-1 | Low | Normal |
| Gastric mucosa | Body/fundus atrophy, antrum spared (Type A gastritis) | Normal |
PA is fundamentally a gastric disease. Many patients may not even have anemia initially - early on, iron deficiency anemia can precede megaloblastic anemia because achlorhydria also impairs iron absorption.
Hematologic Presentation
Both cause megaloblastic anemia when fully established, but:
| Feature | PA | Dietary Deficiency |
|---|---|---|
| Severity at presentation | Often severe - pancytopenia, MCV markedly elevated, marked hypersegmentation | Often mild-moderate - subtle macrocytosis may be the only finding |
| Rate of development | Faster (months to ~2 years once IF is lost) | Slower (years) |
| Presentation without anemia | Common - neurological disease may dominate even with normal CBC | Less common |
| Concomitant iron deficiency | Common (achlorhydria impairs iron absorption) - can mask macrocytosis | Less common |
Neurological Involvement
Both can cause subacute combined degeneration (SCD) of the spinal cord, but this is more prominent and often the presenting feature in PA.
Key neurological points in the comparison:
| Aspect | PA | Dietary Deficiency |
|---|---|---|
| Frequency of SCD | Higher - often presents as the primary or early manifestation | Less common; tends to occur later and only in severe/prolonged deficiency |
| Neurological disease without anemia | Well recognized and common in PA | Less typical |
| Severity | Can progress to paraplegia, cognitive decline ("megaloblastic madness"), optic atrophy | Usually milder paresthesias; severe cases rare except in infants of deficient mothers |
| Reversibility | Incomplete if longstanding | Usually fully reversible with dietary correction |
The spinal cord lesion begins with symmetric paresthesias (usually lower extremities), then loss of vibration sense and proprioception (posterior column), then spasticity and hyperreflexia (lateral column), and in severe cases paraplegia, incontinence, and psychiatric features.
(Goldman-Cecil, p. 1732)
Laboratory Diagnosis
| Test | PA | Dietary Deficiency |
|---|---|---|
| Serum B12 | Low (<74 pmol/L in overt deficiency) | Low |
| MMA | Elevated | Elevated (if severe) |
| Homocysteine | Elevated | Elevated (if severe) |
| Anti-IF antibodies | Positive in 50-70% (highly specific) | Negative |
| Anti-parietal cell Ab | Positive in 90% (less specific) | Negative |
| Fasting serum gastrin | Elevated | Normal |
| Pepsinogen-1 | Low | Normal |
| Holotranscobalamin | Low | Low |
| False-normal serum B12? | Yes - anti-IF antibodies in serum cause false-normal in up to 50% of CBLA assays | No |
| Co-existing iron deficiency | Common | Less common |
The Schilling test (radiolabeled B12 absorption) is no longer available. The diagnostic workup for PA now relies on anti-IF antibodies + elevated gastrin + low pepsinogen-1 + endoscopic biopsy.
Treatment: Critical Differences
| Aspect | PA | Dietary Deficiency |
|---|---|---|
| Duration of treatment | Lifelong - the gastric disease is permanent and progressive | Until dietary correction is established; finite course |
| Route (first-line) | IM/SC cyanocobalamin or hydroxocobalamin 1,000 µg (loading regimen then monthly) | Oral supplementation or dietary change |
| Can high-dose oral work in PA? | Yes - 1,000-2,000 µg/day uses passive absorption (~1% of dose absorbed without IF); equivalent to IM in clinical trials | Yes, and at lower doses (50-100 µg/day) |
| Preferred injectable | Hydroxocobalamin retained longer; monthly vs. cyanocobalamin | Either works; often not needed |
| Monitoring | Annual B12 levels; periodic endoscopy for gastric cancer surveillance | B12 levels every 1-2 years once on supplementation |
| If folate given alone | Dangerous - corrects anemia but allows neurological damage to progress | Same warning applies |
Summary Table
| Feature | Pernicious Anemia | Dietary B12 Deficiency |
|---|---|---|
| Cause | Autoimmune - loss of IF | Insufficient intake |
| Onset | Faster (months-years) | Slow (years-decades) |
| Typical patient | Older adult, European/African ancestry | Vegan, low-resource diet |
| Autoimmune associations | Yes (thyroid, T1DM, vitiligo, Addison's) | No |
| Gastric cancer risk | Elevated (RR 6.8) | Not elevated |
| Anti-IF antibodies | Present (50-70%) | Absent |
| Achlorhydria/high gastrin | Yes | No |
| Neurological disease | Prominent, may precede anemia | Less prominent, occurs late |
| Iron deficiency | Common (coexists) | Less common |
| Treatment duration | Lifelong | Finite |
| Oral therapy feasible? | Yes (high-dose) | Yes (standard dose) |
| Enterohepatic circulation | Disrupted | Intact |
Sources:
- Goldman-Cecil Medicine International Edition, pp. 1729-1735
- Harrison's Principles of Internal Medicine, 22nd ed. (2025), pp. 824-833
- Henry's Clinical Diagnosis and Management by Laboratory Methods, pp. 1243-1245
- Harper's Illustrated Biochemistry, 32nd ed., p. 556
Paget disease
Finding Sources
Finding Sources
Searching PubMed
"Paget disease" AND bone
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
I now have comprehensive content. Let me compile the full answer.
Paget Disease of Bone (Osteitis Deformans)
Definition
Paget disease of bone is a focal, chronic disorder of skeletal remodeling characterized by localized areas of markedly increased and disorganized bone turnover. Overactive osteoclasts initiate excessive bone resorption, followed by a compensatory but structurally chaotic osteoblastic response - producing bone that is enlarged, hypervascular, and mechanically weak.
(Goldman-Cecil Medicine; Harrison's Principles of Internal Medicine, 22nd ed.)
Epidemiology
| Feature | Detail |
|---|---|
| Prevalence | ~0.5-3% in the UK/US; ~3% at autopsy in those >40 years |
| Sex | Males > Females |
| Age | Rarely diagnosed before age 50; increases steeply with age |
| Geography | Common in Western Europe (UK, France, Germany - NOT Switzerland or Scandinavia), and descendants of European migrants to Australia, NZ, South Africa, Americas |
| Race | Rare in native populations of Africa, Asia, Middle East, and the Americas |
| Trend | Prevalence and severity are decreasing and age of diagnosis is increasing - possibly linked to measles vaccination |
Radiographic prevalence in those >55 years: 2.5% in men, 1.6% in women.
Etiology - Genetic and Viral
The etiology remains incompletely understood, but both genetic and viral factors are implicated.
Genetic Factors
- Positive family history in 15-25% of patients; raises prevalence 7-10 fold among first-degree relatives
- SQSTM1 gene (sequestosome-1/p62 protein) - the most common mutation in both familial and sporadic cases; mutations in the C-terminal ubiquitin-binding domain
- TNFRSF11A (encodes RANK) - mutations cause familial expansile osteolysis and early-onset Paget's
- TNFRSF11B (encodes osteoprotegerin/OPG) - homozygous deletion causes juvenile Paget's (familial idiopathic hyperphosphatasia)
- Other candidate genes: CSF1 (M-CSF), RIN3, OPTN, TM7SF4 (DC-STAMP, needed for osteoclast fusion)
- VCP mutations → inclusion body myopathy with Paget's disease and frontotemporal dementia (IBMPFD)
Viral Hypothesis
- Paramyxovirus-like inclusions (resembling measles, respiratory syncytial virus, canine distemper virus) in pagetic osteoclast nuclei and cytoplasm
- Vectors containing measles virus nucleocapsid genes convert osteoclast precursors into pagetic-like giant osteoclasts
- Decline in Paget's incidence coincides with widespread measles vaccination
- However: live virus cannot be cultured from pagetic bone; full-length viral genes cannot be cloned; antibody levels against paramyxoviruses are NOT elevated in Paget's patients
(Harrison's, pp. 3354-3355)
Pathophysiology
The driving cellular abnormality is in the osteoclast.
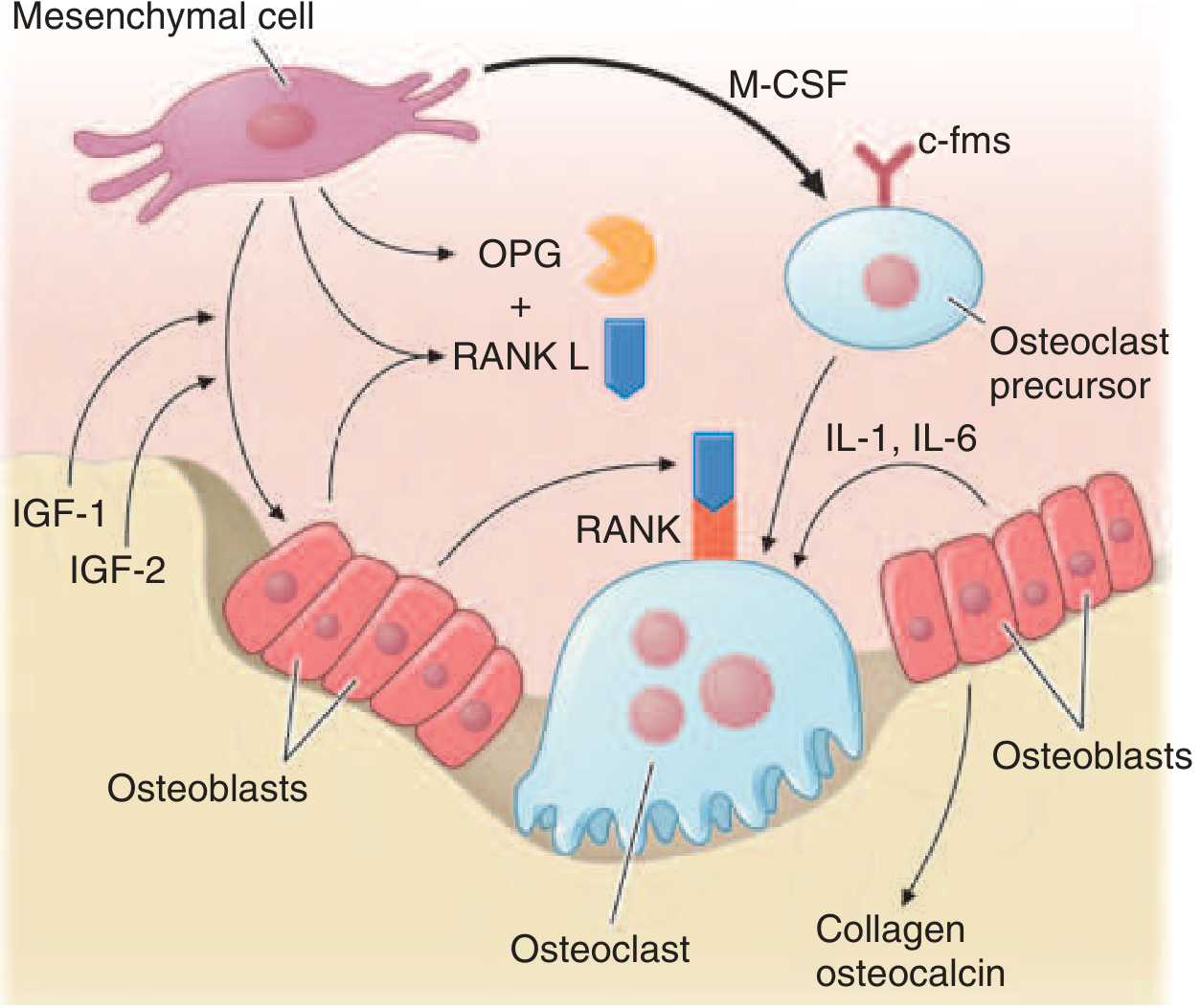
Pagetic Osteoclasts Are Pathologically Abnormal:
- 10-100× increased in number
- Giant cells with up to 100 nuclei (normal osteoclast: 3-5 nuclei)
- Create a sevenfold increase in resorptive surfaces
- Erosion rate of 9 µg/day (normal: 1 µg/day)
Mechanisms of Osteoclast Overactivation:
- Hypersensitivity to 1,25(OH)₂D₃ (active vitamin D)
- Hyperresponsiveness to RANKL (osteoclast stimulatory factor)
- Increased RANKL expression by pagetic marrow stromal cells
- Elevated IL-6 in blood and overexpressed in pagetic osteoclasts → increased osteoclast precursor recruitment
- Upregulation of c-fos proto-oncogene → increased osteoclastic activity
- Overexpression of antiapoptotic Bcl-2 → osteoclast survival prolonged
Three Histological Phases:
| Phase | Characteristics | Radiographic appearance |
|---|---|---|
| Lytic (hot) phase | Osteoclast-dominant; prominent bone resorption; marked hypervascularization | "Blade of grass" / advancing lytic wedge |
| Mixed phase | Active bone formation AND resorption; woven bone replaces lamellar bone; fibrous tissue replaces marrow | Mixed lytic/sclerotic; coarsened trabeculae |
| Sclerotic (burnt-out) phase | Osteoblast-dominant; dense but architecturally chaotic bone; decreased vascularity | Dense, enlarged, sclerotic bone |
Key point: Bone mass is normal or increased (not decreased) in Paget's, unlike osteoporosis. But the bone is structurally weak because of its disorganized mosaic pattern.
(Harrison's, pp. 3355-3356)
Sites of Involvement
Most common sites (in decreasing order):
- Pelvis (most common - ~70%)
- Vertebral bodies (lumbar > thoracic > cervical)
- Skull
- Femur
- Tibia
The disease is polyostotic (multiple bones) in ~65% and monostotic (one bone) in ~35%. It does NOT involve the hands, feet, or ribs commonly.
Clinical Manifestations
Most patients are asymptomatic - diagnosed incidentally via elevated ALP or incidental radiograph. Among symptomatic patients:
Bone and Joint
- Bone pain - most common symptom; due to hypervascularization, lytic lesions, fractures, or periosteal stretching
- Bowing deformity - femur and tibia; produces a characteristic "saber shin" (anterior tibial bowing)
- Secondary osteoarthritis - of hip or knee adjacent to pagetic long bones
- Fractures - usually in long bones at lytic fronts; femoral shaft and subtrochanteric region are most common sites; characteristic "banana fracture" (transverse fracture through a bowed bone)
- Leg-length discrepancy from long bone deformity
Skull / Neurological
- Hearing loss - most common neurological complication; sensorineural (cochlear nerve compression from temporal bone involvement) and/or conductive
- Frontal bossing - symmetric or asymmetric enlargement of parietal and frontal bones; increased hat size (classic finding)
- Cranial nerve palsies from narrowed foramina
- Platybasia (softening of skull base) → risk of brainstem compression
- Headache
- Facial deformity, dental problems, airway compression (rare)
Spinal
- Back pain from enlarged pagetic vertebrae, vertebral compression fractures, spinal stenosis
- Kyphosis and forward tilt
- Spinal cord compression (rare) - from bone enlargement or vascular steal
Cardiovascular
- With involvement of 15-35% of the skeleton and very active disease (ALP ≥4× normal): extensive arteriovenous shunting → high-output cardiac state
- High-output heart failure is rare and usually requires concomitant cardiac disease
- Calcific aortic stenosis and diffuse vascular calcifications are associated
Malignant Transformation
- Osteosarcoma - most common; usually presents as new or worsening pain in a longstanding pagetic lesion; incidence <0.5% (rare but important)
- Incidence is decreasing, possibly due to earlier bisphosphonate therapy
- Benign giant cell tumors (osteoclast-rich) may arise adjacent to pagetic bone; respond to glucocorticoids
Diagnosis
Biochemical Markers
| Marker | Finding | Notes |
|---|---|---|
| Serum ALP | Markedly elevated | Best single marker of disease activity and treatment response; ALP correlates with extent and activity |
| Bone-specific ALP | More specific | Preferred if liver disease present |
| Serum calcium/phosphate | Normal | Hypercalcemia may occur during immobilization; hypocalcemia may occur with very active disease + poor calcium intake during bisphosphonate therapy |
| Urinary hydroxyproline | Elevated | Less used now |
| Serum/urine N-telopeptide, C-telopeptide | Elevated | Bone resorption markers |
Radiology - Classic X-ray Signs:
| Region | Radiographic Sign |
|---|---|
| Long bones (lytic phase) | "Blade of grass" - advancing V-shaped lytic front |
| Skull | "Cotton wool" appearance (mixed lytic/sclerotic); "osteoporosis circumscripta" (pure lytic); thickened diploë; enlargement of skull bones |
| Vertebra | "Picture frame vertebra" (cortical thickening of end plates); "Ivory vertebra" (diffuse radiodense enlargement) |
| Pelvis | Thickened iliopectineal line (brim sign); coarsened trabeculae |
| Long bones (sclerotic) | Cortical thickening, bowing, coarsened trabeculae |
Imaging Modalities:
- Radionuclide bone scan - most sensitive test; best for defining the full extent of disease (all affected sites)
- Plain X-ray - diagnostic for typical features; used to characterize specific lesions
- CT/MRI - used for spinal stenosis, cord compression, and sarcoma evaluation
(Harrison's, pp. 3356-3357; Goldman-Cecil, p. 2651)
Treatment
Indications for Pharmacologic Therapy (Endocrine Society 2014 Guidelines):
- Bone pain localized to a pagetic site
- Before surgery at an active pagetic site (to reduce intraoperative bleeding)
- To reduce hypercalciuria during immobilization
- High disease activity (elevated ALP) with involvement of weight-bearing bones, areas near joints, vertebral bodies, or skull
- Neurological complications
Note: Current evidence (UK randomized trials) shows no difference in bone pain, fracture rates, or quality of life between symptom-directed and ALP-normalizing bisphosphonate strategies - so asymptomatic patients do not routinely need treatment to normalize ALP alone.
Drug Therapy:
| Drug | Regimen | ALP Normalization |
|---|---|---|
| Zoledronic acid (first-line) | 5 mg IV over 15 min (single dose) | 90% at 6 months |
| Risedronate | 30 mg/day PO × 2 months | 73% |
| Alendronate | 40 mg/day PO × 6 months | 63% |
| Pamidronate | 30 mg/day IV × 3 days | ~50% |
| Tiludronate | 800 mg/day PO × 3 months | 35% |
| Etidronate | 200-400 mg/day PO × 6 months | 15% |
| Calcitonin (salmon) | 100 U SC daily × 6-18 months | Reduces ALP ~50% (not normalization) |
Zoledronic acid is the treatment of choice - single infusion, prolonged duration of action lasting several years, highest remission rate.
Important Precautions:
- Always ensure adequate calcium and vitamin D intake before bisphosphonate therapy - active bone formation continues while resorption is suppressed, risking hypocalcemia
- Monitor ALP every 3-6 months to assess response
- Hearing loss from temporal bone involvement may not respond to bisphosphonates
Summary: Key "Exam Facts"
| Feature | Detail |
|---|---|
| Core mechanism | Overactive osteoclasts → disorganized bone remodeling |
| Most common symptom | Bone pain |
| Most common complication | Bone deformity (bowing) |
| Most common neurological complication | Deafness (temporal bone involvement) |
| Key biochemical marker | Elevated serum ALP (Ca/P normal) |
| Most sensitive imaging | Radionuclide bone scan |
| Pathognomonic X-ray signs | "Blade of grass," "cotton wool," "picture frame vertebra," "ivory vertebra" |
| Feared malignant complication | Osteosarcoma (<0.5%) |
| First-line treatment | Zoledronic acid 5 mg IV (single dose) |
| Autosomal dominant gene (sporadic + familial) | SQSTM1 (p62/sequestosome-1) |
Sources:
- Harrison's Principles of Internal Medicine, 22nd Edition (2025), pp. 3354-3358
- Goldman-Cecil Medicine International Edition, pp. 2649-2652
- Katzung's Basic and Clinical Pharmacology, 16th Edition
- Rockwood and Green's Fractures in Adults, 10th Edition (2025)
Multiple myeloma
Finding Sources
Searching PubMed
"multiple myeloma" AND treatment
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from Harrison's, Goldman-Cecil, and Robbins to compile a thorough answer.
Multiple Myeloma
Definition
Multiple myeloma (MM) is a malignancy of clonal plasma cells that infiltrate the bone marrow and produce a monoclonal immunoglobulin (M protein or paraprotein). The tumor, its products, and the host response result in a constellation of end-organ damage: bone destruction, renal failure, anemia, immune suppression, and hypercalcemia.
Formally defined by:
- ≥10% clonal plasma cells on bone marrow examination
- M protein in serum or urine (except nonsecretory MM)
- One or more myeloma-defining events (CRAB criteria or biomarkers - see below)
(Goldman-Cecil Medicine; Harrison's Principles of Internal Medicine, 22nd ed.)
Epidemiology
| Parameter | Data |
|---|---|
| US incidence (2024) | ~35,780 new cases/year; ~12,540 deaths |
| % of all malignancies | ~1.8% |
| % of hematologic malignancies | ~10% |
| Median age at diagnosis | 69 years |
| Age <40 | Uncommon (<2%) |
| Sex | Males > Females |
| Race | Blacks have nearly twice the incidence of Whites |
| Incidence (per 100,000) | White men 8.1, White women 5.1; Black men 17.1, Black women 13.0 |
MM is the second most common hematologic malignancy after non-Hodgkin lymphoma.
The Plasma Cell Spectrum: MGUS → Smoldering MM → Active MM
Almost all cases of MM evolve from a premalignant MGUS (Monoclonal Gammopathy of Undetermined Significance) phase. MGUS is clinically recognized before myeloma in only a minority of patients. The risk of MGUS progressing to MM is approximately 1% per year.
MGUS Risk of Progression to Myeloma (20-year risk):
| Risk Group | Criteria | 20-yr cumulative risk |
|---|---|---|
| Low risk | M protein <1.5 g/dL, IgG subtype, normal FLC ratio (0.26-1.65) | 5% |
| Low-intermediate | Any one factor abnormal | 21% |
| High-intermediate | Any two factors abnormal | 37% |
| High risk | All three factors abnormal | 58% |
Smoldering Multiple Myeloma (Asymptomatic):
- Serum M protein (IgG or IgA) ≥20 g/L or urine M protein ≥500 mg/24h AND/OR clonal BM cells 10-60%
- No myeloma-defining events or amyloidosis
Symptomatic Multiple Myeloma - CRAB Criteria:
| Letter | Criterion | Threshold |
|---|---|---|
| C | Calcium (hypercalcemia) | >0.25 mmol/L above ULN OR >2.75 mmol/L (11 mg/dL) |
| R | Renal insufficiency | CrCl <40 mL/min OR creatinine >177 µmol/L (>2 mg/dL) |
| A | Anemia | Hb >20 g/L below LLN OR <100 g/L |
| B | Bone lesions | ≥1 osteolytic lesion on X-ray, CT, or PET-CT |
Plus SLiM biomarkers (even without CRAB):
- Sixty percent: clonal BM plasma cells ≥60%
- Light chain ratio: involved:uninvolved serum FLC ratio ≥100
- MRI: >1 focal lesion on MRI
(Harrison's, p. 927-928)
Pathogenesis and Molecular Biology
Origin and Progression
All MM evolves through MGUS via a two-hit model - the risk of progression is approximately constant (~1%/year) regardless of MGUS duration. The two main molecular events driving MGUS→MM progression are RAS mutations and MYC abnormalities. The bone marrow microenvironment plays a major enabling role, particularly through IL-6 (a major plasma cell growth factor produced by marrow fibroblasts and macrophages).
Key Cytogenetic Abnormalities
MM is divided into two main cytogenetic subtypes:
| Subtype | Frequency | Key Features |
|---|---|---|
| Hyperdiploid | ~40% | Trisomies of odd chromosomes (3, 5, 7, 9, 11, 15, 19, 21); generally better prognosis |
| IgH-translocated (non-hyperdiploid) | ~40% | Translocations involving chromosome 14q32 (IgH locus) |
| Both features | ~15% | |
| Other | ~5% |
High-risk IgH translocations:
- t(4;14)(p16;q32) - involves FGFR3/MMSET; adverse
- t(14;16)(q32;q23) - involves MAF; adverse
- t(14;20) - involves MAFB; adverse
Standard-risk translocations:
- t(11;14)(q13;q32) - involves cyclin D1; standard risk (but note: this is present in nearly all AL amyloidosis cases)
Other adverse cytogenetic features:
- del(17p13) - TP53 deletion; very high risk
- del(13q14)
- 1q amplification / 1p deletion
Most frequent somatic mutations: KRAS (~20%), NRAS (~20%), TP53, DIS3, FAM46C, BRAF (each 5-10%).
Bone Disease Mechanism
Myeloma cells produce factors that:
- Upregulate RANKL expression by bone marrow stromal cells → osteoclast activation
- Downregulate OPG (decoy receptor for RANKL) → increased RANKL/OPG ratio
- Inhibit osteoblasts via DKK1 (dickkopf-1), IL-3, IL-7
Result: Pure osteolytic lesions (unlike Paget disease, where both resorption and formation are increased). This is the hallmark of myeloma bone disease. Fractures, hypercalcemia, and bone pain follow.
Other cytokines contributing to osteoclast activation: MIP-1α, SDF-α, IL-1β, IL-6.
(Goldman-Cecil, p. 1978; Robbins Basic Pathology)
M Protein Distribution
| Type | Frequency |
|---|---|
| IgG | 52-60% (most common) |
| IgA | 20-21% |
| Light chain only (Bence Jones) | 16% |
| IgD | 2% |
| Biclonal | 2% |
| Nonsecretory | ~3% |
| Light chain type: κ | 65% |
| Light chain type: λ | 35% |
Clinical Features
Presenting Symptoms
Bone pain (back/chest/extremities) is the most common symptom at diagnosis - present in >2/3 of patients. Vertebral collapse may reduce height by several inches.
| System | Manifestation | Mechanism |
|---|---|---|
| Skeletal | Bone pain, pathologic fractures, vertebral collapse | Osteolysis via RANKL/OPG imbalance |
| Hematologic | Normocytic normochromic anemia (75% at presentation; nearly 100% eventually) | BM infiltration, cytokine suppression of erythropoiesis |
| Renal | Renal insufficiency (cast nephropathy most common) | Bence Jones proteins form obstructive casts in distal tubules and collecting ducts |
| Immune | Recurrent bacterial infections | Hypogammaglobulinemia (functional Ab suppressed despite high total Ig), neutropenia |
| Metabolic | Hypercalcemia | Osteolysis + immobilization |
| Neurological | Radiculopathy (most common CNS complication - thoracic/lumbosacral) | Vertebral compression, nerve root entrapment |
| Spinal cord compression | Up to 10% of patients | Vertebral collapse/epidural extension |
| Hyperviscosity | Headache, visual disturbance, confusion | High M protein (especially IgA/IgM) |
| Coagulation | DVT (also caused by lenalidomide therapy) | Multiple mechanisms |
| Amyloidosis | ~10% develop AL amyloidosis | Light chain deposition |
Physical Examination
- Pallor (most frequent finding)
- Hepatomegaly (~5%), splenomegaly (~1%)
- Bone tenderness
- Palpable extramedullary plasmacytomas
Diagnosis
Serum Protein Electrophoresis (SPEP) and Immunofixation
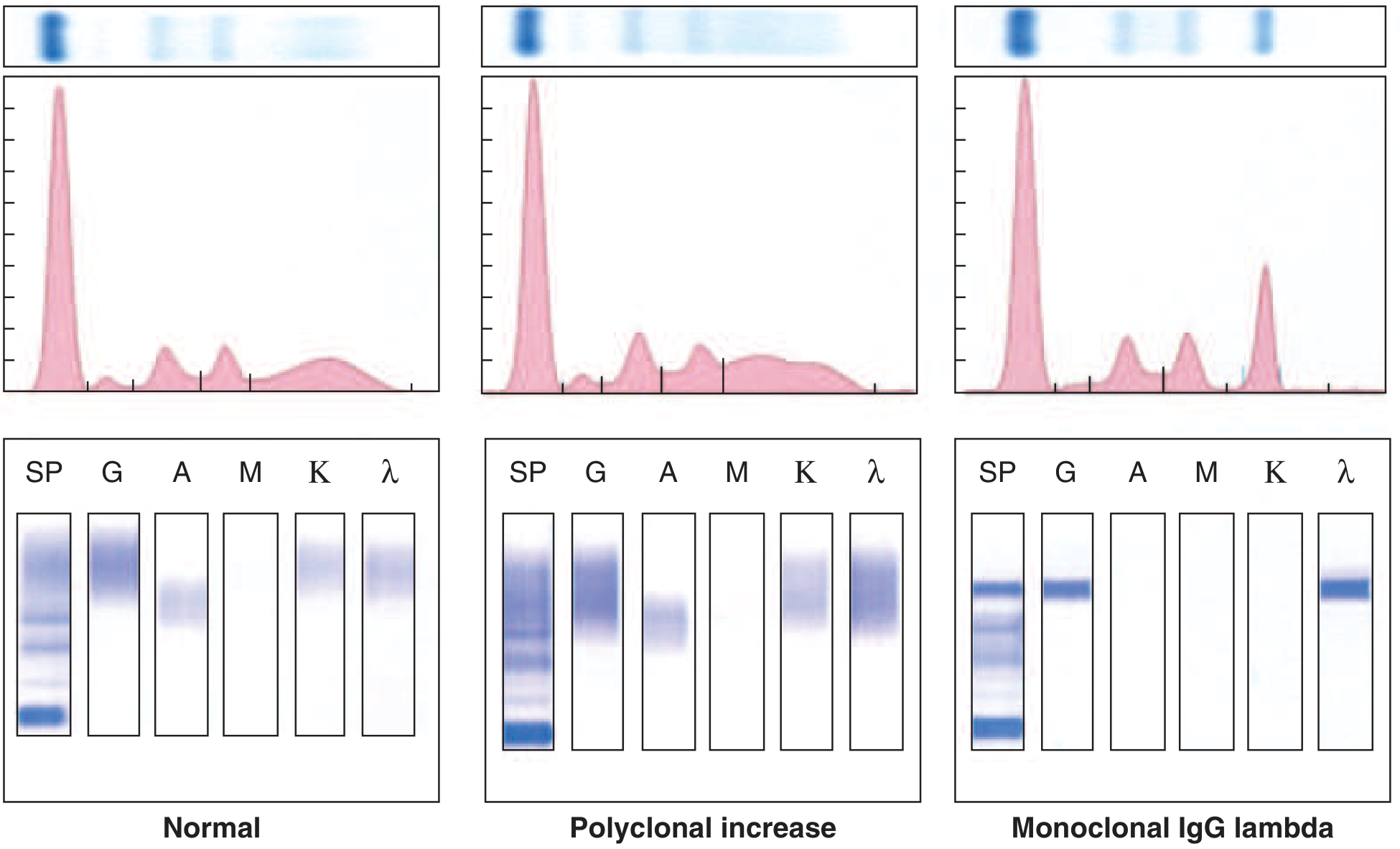
| Test | Finding | Sensitivity |
|---|---|---|
| SPEP alone | M protein | 80% |
| SPEP + serum immunofixation | M protein | 93% |
| SPEP + serum immunofixation + urine immunofixation | M protein | 97% |
| Serum free light chain (FLC) assay | Elevated involved:uninvolved FLC ratio | Can replace urine studies |
Bone Marrow Examination
- Clonal plasma cells >10% in 96% of patients
- Plasma cell immunophenotype: CD138+, CD38+, CD45−, CD19−, CD56+; cytoplasmic Ig+
- Clonality confirmed by κ/λ ratio: >4:1 (κ clonal) or <1:2 (λ clonal)
Imaging
- Whole-body low-dose CT or PET-CT: preferred initial imaging (replaces skeletal survey)
- MRI: most sensitive for spinal involvement, cord compression, marrow infiltration
- PET-CT: excellent for extramedullary disease and treatment response
- Classic X-ray: "punched-out" lytic lesions (1-4 cm), most commonly in skull, spine, ribs, pelvis, femur
Other Lab Findings
- Anemia (normocytic, normochromic) - most common lab abnormality
- Elevated creatinine (~25% at diagnosis)
- Elevated calcium
- Elevated LDH and β₂-microglobulin (prognostic markers)
- Elevated serum protein with low albumin
- Rouleaux formation on peripheral blood smear
Staging
Revised International Staging System (R-ISS):
| Stage | Criteria | Median OS |
|---|---|---|
| I | β₂M <3.5 mg/L AND albumin ≥3.5 g/dL AND no high-risk cytogenetics AND normal LDH | ~Not reached (>5 years) |
| II | Neither I nor III | ~83 months |
| III | β₂M ≥5.5 mg/L AND (high-risk cytogenetics OR elevated LDH) | ~43 months |
High-risk cytogenetics for R-ISS: del(17p), t(4;14), t(14;16)
Treatment
Transplant-Eligible Patients (~50% of newly diagnosed MM)
Good performance status, limited comorbidities, physiologic age <65-70 years.
Standard induction (3-4 months):
- VRd: Bortezomib + Lenalidomide + Dexamethasone (backbone)
- DaraVRd (Daratumumab + VRd): for high-risk disease; superior outcomes in recent trials
- VCD: Bortezomib + Cyclophosphamide + Dexamethasone (alternative)
- DRd: Daratumumab + Lenalidomide + Dexamethasone (alternative)
Stem Cell Transplantation:
- Peripheral blood stem cells mobilized with G-CSF ± plerixafor ± cyclophosphamide
- Conditioning: Melphalan 200 mg/m² (high-dose) then autologous SCT
- Not curative, but prolongs event-free and overall survival vs. conventional chemotherapy
- Collect enough cells for 1-2 transplants
Maintenance: Lenalidomide (standard post-ASCT maintenance, given indefinitely or until progression)
Transplant-Ineligible Patients
- DaraRd (Daratumumab + Lenalidomide + Dexamethasone): current standard of care
- VRd (lower-intensity version with bortezomib once weekly)
- VMP: Bortezomib + Melphalan + Prednisone (older regimen, still used in some settings)
Smoldering MM:
- Low/intermediate risk: observation every 3-4 months; no treatment until progression
- High-risk smoldering: Lenalidomide ± dexamethasone for ~2 years significantly reduces progression to active MM and overall mortality
Relapsed/Refractory MM:
Multiple lines of therapy available. Key agents and classes:
| Drug Class | Examples | Key Notes |
|---|---|---|
| Proteasome inhibitors | Bortezomib (IV/SC), Carfilzomib, Ixazomib (oral) | Bortezomib causes peripheral neuropathy |
| IMiDs (immunomodulatory) | Thalidomide, Lenalidomide, Pomalidomide | DVT risk; require prophylactic anticoagulation |
| Anti-CD38 monoclonal Ab | Daratumumab, Isatuximab | Major advance; now used frontline |
| Anti-SLAMF7 | Elotuzumab | Used in combination |
| BCL-2 inhibitor | Venetoclax | Particularly active in t(11;14) myeloma |
| Nuclear export inhibitor | Selinexor | Refractory disease |
| Alkylating agents | Melphalan, Cyclophosphamide, Bendamustine | |
| Steroids | Dexamethasone | Partner for all regimens |
| CAR-T cell therapy | Ciltacabtagene autoleucel (cilta-cel), Idecabtagene vicleucel (ide-cel) - target BCMA | Transformative for heavily pretreated MM; high response rates |
| Bispecific antibodies | Teclistamab (anti-BCMA × CD3), Elranatamab, Talquetamab (anti-GPRC5D) | Outpatient option for relapsed/refractory |
2025 NCCN Update (PMID 40340857): Current guidelines continue to refine frontline quadruplet therapies and the role of MRD-guided treatment decisions. The NCCN Multiple Myeloma v1.2025 guidelines represent the current standard.
High-risk MM consensus (PMID 40489728): The IMS/IMWG 2025 consensus on defining high-risk MM recommends specific cytogenetic and genomic criteria to guide more aggressive frontline therapy.
Supportive Care
| Complication | Management |
|---|---|
| Bone disease | Bisphosphonates (zoledronic acid or pamidronate monthly) - reduce skeletal events; denosumab is an alternative |
| Hypercalcemia | IV fluids, bisphosphonates, calcitonin, steroids |
| Renal failure | Aggressive hydration, avoid NSAIDs/contrast, plasmapheresis for hyperviscosity, dose-adjust renally cleared drugs |
| Anemia | Erythropoiesis-stimulating agents (ESA), transfusion |
| Infections | Prophylactic antivirals (acyclovir with bortezomib); PCP prophylaxis; IVIG for recurrent infections; vaccinations (avoid live vaccines) |
| DVT prophylaxis | Aspirin or anticoagulation (especially with lenalidomide-based regimens) |
| Pain/fractures | Radiation therapy for localized bone lesions; orthopedic stabilization; kyphoplasty/vertebroplasty for vertebral fractures |
| Hyperviscosity | Plasmapheresis |
Prognosis
- Median OS has improved dramatically from ~2-3 years (pre-novel agent era) to 7-10+ years with modern therapy for standard-risk disease
- High-risk MM (del17p, t(4;14), t(14;16), 1q gain) still has median OS of ~3-4 years
- Minimal residual disease (MRD) negativity by flow cytometry or next-generation sequencing is the strongest predictor of long-term outcomes
- MM remains incurable with current therapy, but CAR-T and bispecific antibodies are producing deep, prolonged remissions
Key "Exam Facts"
| Feature | Detail |
|---|---|
| Cell of origin | Clonal plasma cell |
| Most common M protein | IgG (52-60%) |
| Most common presenting symptom | Bone pain |
| Most common lab abnormality | Anemia (normocytic, normochromic) |
| Bone lesion type | Punched-out lytic lesions (no sclerosis, no osteoblast activity) |
| Bone lesion mechanism | RANKL↑ / OPG↓ → osteoclast activation + osteoblast inhibition (DKK1) |
| Bence Jones protein | Free light chains (κ or λ) in urine |
| SPEP pattern | "Church spire" M-spike in γ-region |
| Renal injury mechanism | Myeloma cast nephropathy (Bence Jones proteins in distal tubules) |
| Diagnostic cutoff | BM plasma cells ≥10% |
| Best initial imaging | Whole-body low-dose CT or PET-CT |
| Staging | R-ISS (uses β₂M, albumin, LDH, cytogenetics) |
| Induction (transplant eligible) | VRd ± Daratumumab |
| Conditioning for ASCT | Melphalan 200 mg/m² |
| Most potent current agents | Anti-CD38 mAbs (daratumumab); proteasome inhibitors; CAR-T (BCMA-targeted) |
| Bone protection | Zoledronic acid monthly |
| Most common neurologic complication | Radiculopathy (vertebral compression) |
| Feared but rare complication | Plasma cell leukemia (peripheral blood plasmacytosis) |
Sources:
- Harrison's Principles of Internal Medicine, 22nd Edition (2025), pp. 927-932
- Goldman-Cecil Medicine International Edition, pp. 1977-1983
- Robbins & Kumar Basic Pathology, pp. 2871-2884
- NCCN Guidelines - Multiple Myeloma v1.2025 (PMID 40340857)
- IMS/IMWG High-Risk MM Consensus 2025 (PMID 40489728)
This is a shared conversation. Sign in to Orris to start your own chat.