Diabetes mellitus
Reading File
Finding Sources
Searching PubMed
"diabetes mellitus" AND management
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Searching the Web
diabetes mellitus ADA 2025 2026 management guidelines updates
I now have extensive, authoritative content from Robbins and Harrison's 22e. Let me compile the comprehensive answer.
Diabetes Mellitus
Diabetes mellitus (DM) is a group of metabolic disorders sharing the common feature of hyperglycemia, caused by defects in insulin secretion, insulin action, or most commonly both. Chronic hyperglycemia produces secondary damage in the kidneys, eyes, nerves, and blood vessels. In the United States it is the leading cause of end-stage renal disease, adult-onset blindness, and non-traumatic lower-extremity amputations. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p.1014
Epidemiology
-
30 million Americans (>11% of the population) have DM; ~1.9 million have Type 1
- Nearly 25% of affected individuals are unaware of their diagnosis
- ~96 million US adults have prediabetes
- ~422 million people affected worldwide (WHO estimate)
- T2D in adolescents has increased dramatically due to rising childhood obesity
- Total yearly US cost: ~$327 billion ($237B direct medical + $90B indirect)
Pancreatic Islet Histology
The islets of Langerhans contain three main hormone-producing cell types, as shown below:
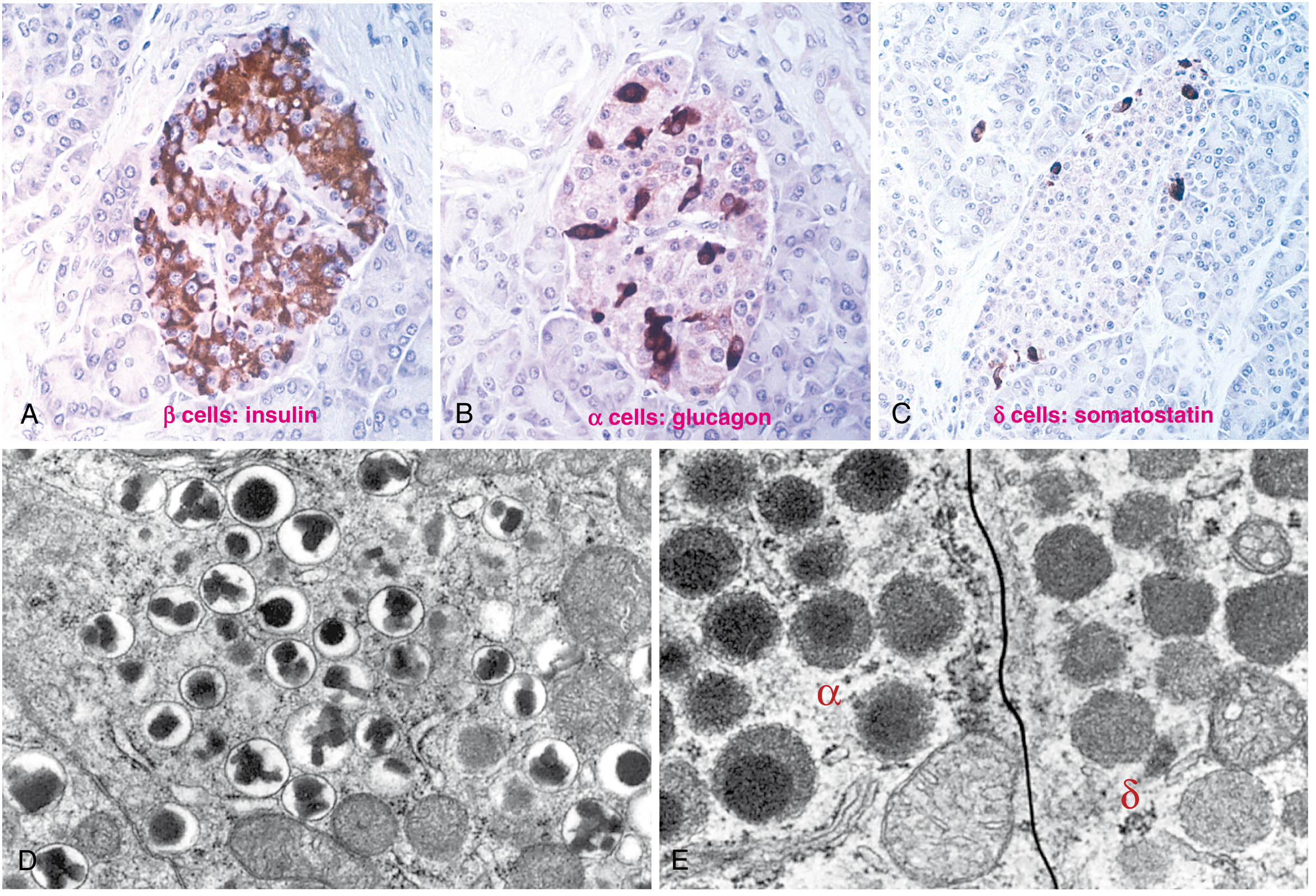
Immunoperoxidase staining of pancreatic islets showing β cells (insulin), α cells (glucagon), and δ cells (somatostatin), with electron micrographs of their distinct secretory granules. (Robbins, Fig. 24.27)
Diagnosis
Blood glucose is normally maintained at 70-120 mg/dL. ADA/WHO diagnostic criteria (any one of the following, confirmed on a separate day except for criterion 2):
| Criterion | Value |
|---|---|
| Fasting plasma glucose | ≥ 126 mg/dL |
| Random plasma glucose + classic symptoms | ≥ 200 mg/dL |
| 2-hr plasma glucose (75g OGTT) | ≥ 200 mg/dL |
| HbA1c | ≥ 6.5% |
Prediabetes criteria (one or more):
- Fasting glucose: 100-125 mg/dL ("impaired fasting glucose")
- 2-hr OGTT glucose: 140-199 mg/dL ("impaired glucose tolerance")
- HbA1c: 5.7-6.4%
Up to one-quarter of individuals with impaired glucose tolerance will develop overt diabetes within 5 years. Even prediabetes carries significant cardiovascular risk. - Robbins, p.1015
Classification
| Feature | Type 1 (T1D) | Type 2 (T2D) |
|---|---|---|
| Prevalence | ~5-10% of DM | ~90-95% of DM |
| Mechanism | Autoimmune β-cell destruction → absolute insulin deficiency | Insulin resistance + relative β-cell insufficiency |
| Age at onset | Usually <20 yrs (can be any age) | Classically adult-onset; now also in youth |
| Body habitus | Often lean | Usually overweight/obese |
| HLA association | Yes (HLA-DR3, DR4) | No |
| Autoantibodies | Yes (anti-GAD, anti-islet cell, anti-insulin) | No |
| Ketoacidosis risk | High | Low (but possible) |
Other categories include: gestational DM, MODY (maturity-onset diabetes of the young), secondary DM (pancreatitis, Cushing syndrome, acromegaly, drugs), and rare monogenic forms.
Pathogenesis
Type 1 Diabetes
T1D involves failure of self-tolerance in T cells specific for islet antigens:
- Autoreactive T cells escape thymic deletion (or regulatory T-cell suppression fails)
- Initial activation occurs in peripancreatic lymph nodes in response to released islet antigens
- Activated Th1 cells secrete IFN-γ and TNF → β-cell injury
- CD8+ cytotoxic T lymphocytes directly kill β cells
- Target autoantigens include insulin, GAD (glutamic acid decarboxylase), and others
- Circulating autoantibodies are markers, not the primary effectors
Three stages of T1D (as recognized by JDRF/ADA):
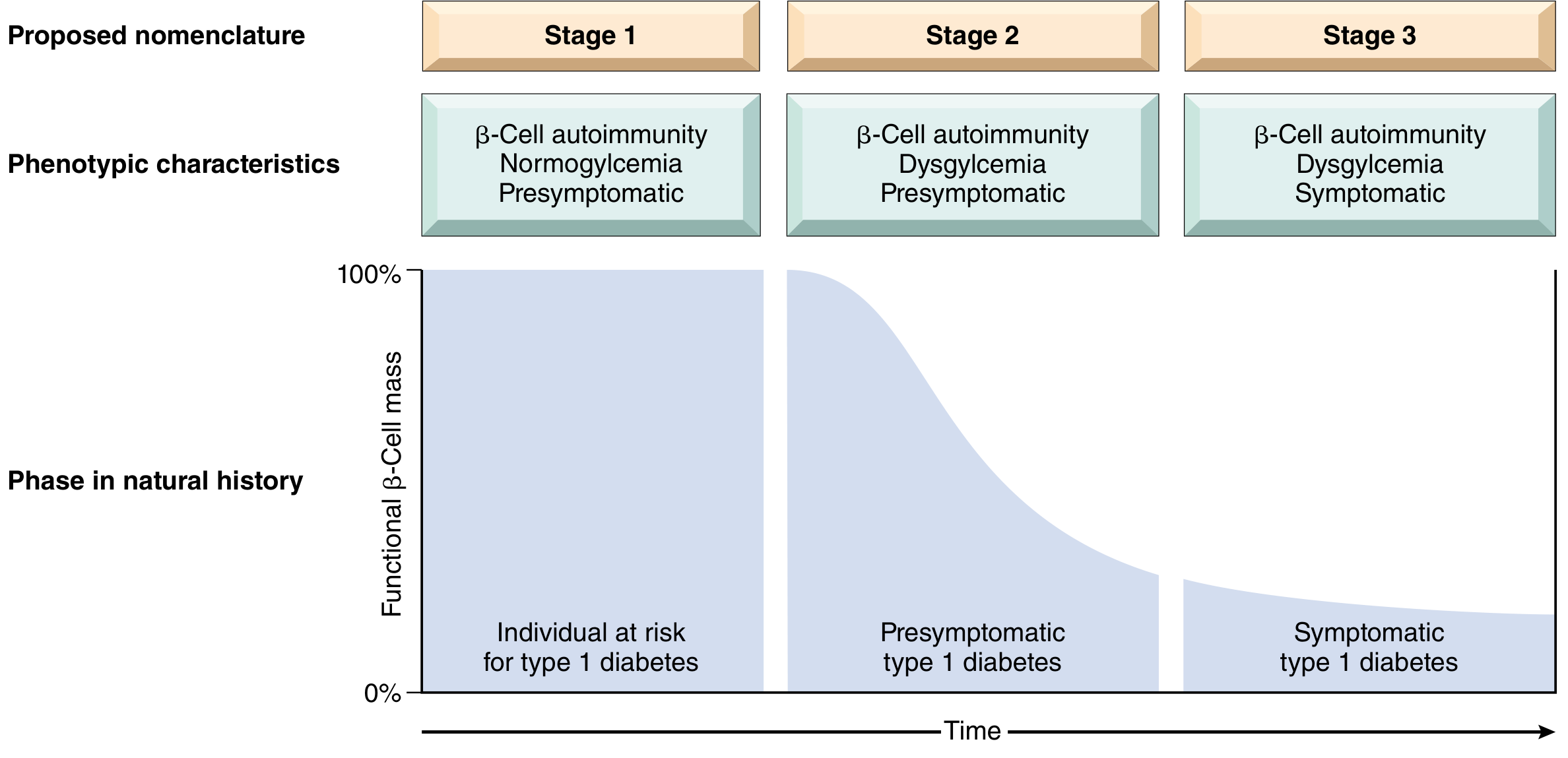
Robbins, Fig. 24.31 - The three stages of T1D, based on the JDRF/Endocrine Society/ADA scientific statement.
- Stage 1: ≥2 islet autoantibodies present; normoglycemia; presymptomatic
- Stage 2: Dysglycemia (impaired glucose tolerance) but still presymptomatic; 75% risk of symptomatic disease within 5 years
- Stage 3: Classic symptomatic disease (polyuria, polydipsia, polyphagia, ketoacidosis) - occurs after >90% β-cell loss
Genetic factors: HLA-DR3 and HLA-DR4 confer the highest risk; monozygotic twin concordance is only ~50%, indicating environmental triggers (viral infections, molecular mimicry with islet antigens).
Type 2 Diabetes
Two central pathogenic defects:
- Peripheral insulin resistance - especially in skeletal muscle, liver, adipose tissue
- β-Cell dysfunction - relative insulin deficiency; β cells cannot compensate for insulin resistance
Obesity drives insulin resistance through:
- Excess free fatty acids (FFAs)
- Pro-inflammatory adipocytokines (e.g., TNF-α, IL-6, leptin, reduced adiponectin)
- Chronic low-grade inflammation
The natural history of T2D: compensatory hyperinsulinemia → impaired glucose tolerance → progressive β-cell exhaustion → overt hyperglycemia.
Clinical Features (Symptoms of Hyperglycemia)
Per Harrison's 22e:
- Polyuria (osmotic diuresis from glucosuria)
- Polydipsia (secondary to polyuria)
- Weight loss (caloric loss in urine + muscle catabolism)
- Fatigue and weakness
- Blurred vision (changes in lens water content - resolves with glycemic control)
- Recurrent infections (vaginitis, fungal skin infections, UTIs)
- Slow wound healing
T2D patients may be asymptomatic for years and present with established chronic complications at diagnosis.
Chronic Complications
Sustained hyperglycemia leads to long-term complications through four key biochemical mechanisms:
| Mechanism | Effect |
|---|---|
| Advanced glycation end-products (AGEs) | Cross-link proteins, activate receptors → vascular damage |
| Protein kinase C (PKC) activation | Alters vascular permeability, gene expression |
| Polyol pathway disturbances | Sorbitol accumulation → osmotic injury (lens, nerves) |
| Hexosamine pathway overload | Alters gene transcription |
Macrovascular (Macroangiopathy)
- Atherosclerosis (accelerated) - coronary, cerebral, peripheral arteries
- Ischemic heart disease (leading cause of death in DM)
- Stroke
- Peripheral artery disease → lower-extremity ischemia, gangrene, amputation
- Women with DM have a sixfold greater risk of dying from CVD vs. non-diabetic women - Harrison's 22e, p.3209
Microvascular (Microangiopathy)
- Diabetic retinopathy - leading cause of adult-onset blindness in the US
- Background (non-proliferative): microaneurysms, hard exudates, flame hemorrhages
- Proliferative: neovascularization → vitreous hemorrhage, retinal detachment
- Diabetic nephropathy - leading cause of end-stage renal disease in the US
- Glomerular hyperfiltration → microalbuminuria → overt proteinuria → declining GFR
- Kimmelstiel-Wilson nodules (nodular glomerulosclerosis) pathognomonic
- Diabetic neuropathy - most common long-term complication
- Distal symmetric polyneuropathy (glove-and-stocking)
- Autonomic neuropathy: gastroparesis, orthostatic hypotension, erectile dysfunction, cardiac denervation
Other Complications
- Diabetic foot - combination of neuropathy + ischemia + infection
- Increased susceptibility to infections (neutrophil dysfunction)
- Skin: acanthosis nigricans, necrobiosis lipoidica, diabetic dermopathy
Acute Complications
| Complication | Type | Mechanism | Key Features |
|---|---|---|---|
| DKA (Diabetic Ketoacidosis) | T1D (mainly) | Absolute insulin deficiency → lipolysis → ketogenesis | pH <7.3, bicarbonate <15, ketonemia, anion gap acidosis, fruity breath |
| HHS (Hyperosmolar Hyperglycemic State) | T2D (mainly) | Relative insulin deficiency, profound dehydration | Glucose often >600 mg/dL, serum osmolality >320 mOsm/kg, minimal ketosis |
| Hypoglycemia | Both | Excess insulin, missed meals, exercise | Glucose <70 mg/dL; sweating, tremor, confusion, seizure |
Management Overview
Glycemic Targets (ADA)
- HbA1c: <7.0% for most adults (individualized; <6.5% in select patients, <8.0% in elderly/high-risk)
- Fasting glucose: 80-130 mg/dL
- Post-prandial peak: <180 mg/dL
Non-Pharmacologic
- Medical nutrition therapy (reduce refined carbohydrates, saturated fat; increase fiber)
- Aerobic + resistance exercise ≥150 min/week
- Weight loss (5-10% body weight improvement significantly improves T2D)
- Smoking cessation
- Sleep hygiene (poor sleep worsens insulin resistance)
Pharmacologic - T1D
- Insulin therapy (mandatory): multiple daily injections or continuous subcutaneous infusion
- Basal insulin (glargine, detemir, degludec) + prandial insulin (lispro, aspart, glulisine)
- Continuous glucose monitoring (CGM)
- Adjunct agents: pramlintide, SGLT2 inhibitors (select patients)
Pharmacologic - T2D (Stepwise)
| Drug Class | Example | Mechanism | Key Benefit |
|---|---|---|---|
| Biguanides | Metformin | Reduces hepatic glucose output | First-line; low cost; weight neutral |
| GLP-1 receptor agonists | Semaglutide, liraglutide | Enhance insulin secretion, suppress glucagon | Weight loss, CV benefit |
| SGLT2 inhibitors | Empagliflozin, dapagliflozin | Block renal glucose reabsorption | CV + renal protection |
| DPP-4 inhibitors | Sitagliptin | Increase GLP-1 activity | Weight neutral |
| Sulfonylureas | Glipizide, glimepiride | Stimulate insulin secretion | Low cost; hypoglycemia risk |
| Thiazolidinediones | Pioglitazone | Improve insulin sensitivity | Fluid retention risk |
| Insulin | Various | Replace deficient insulin | Use when oral agents insufficient |
Current guidance (ADA/ESC): for T2D patients with established ASCVD or CKD, an SGLT2 inhibitor or GLP-1 RA should be added regardless of HbA1c level, given their proven cardiorenal protective effects (class I recommendation).
Monitoring and Screening
Per Harrison's 22e, p.3247-3248:
- Annual dilated retinal exam (ophthalmologist)
- Annual foot examination: pedal pulses, monofilament sensation (10-g), vibratory sensation (128-Hz tuning fork), ankle reflexes
- Annual urine albumin-to-creatinine ratio + eGFR
- Lipid profile at diagnosis and periodically
- Blood pressure at every visit (target <130/80 mmHg)
- Distal symmetric polyneuropathy screening: from diagnosis in T2D; 5 years after diagnosis in T1D
Special Considerations
Sex Differences (Harrison's 22e)
- Women are more sensitive to insulin than men, yet prevalence of T2D is similar between sexes
- Higher testosterone in women → increased T2D risk; lower testosterone in men → increased risk (confirmed by Mendelian randomization)
- PCOS, gestational DM, preeclampsia → significantly elevated T2D risk
- Women with DM lose the "female cardiovascular protection" - their CVD risk equals that of diabetic men
Gestational Diabetes (GDM)
- Defined as glucose intolerance first recognized during pregnancy
- Screened at 24-28 weeks gestation using 50g glucose challenge (then 100g OGTT if abnormal)
- Increases risk of fetal macrosomia, neonatal hypoglycemia, operative delivery
- Mother has high lifetime risk of developing T2D
Summary Pathology Points (Robbins)
T1D: Autoimmune β-cell destruction → absolute insulin deficiency. Circulating autoantibodies to islet antigens (including insulin and GAD). T-cell mediated.
T2D: No autoimmune basis. Insulin resistance + β-cell dysfunction → relative insulin deficiency. Obesity contributes through excess FFAs, pro-inflammatory adipocytokines, and inflammation.
Complications: Both types share identical late complications - macroangiopathy (atherosclerosis, IHD, PAD) and microangiopathy (retinopathy, nephropathy, neuropathy) - all driven by the four biochemical pathways of hyperglycemia-induced damage (AGEs, PKC, polyol, hexosamine).
Recent evidence note: A 2024 meta-analysis (PMID 39037772) confirmed that digital interventions for T2D self-management significantly improve glycemic outcomes. An ongoing meta-analysis (2025) identified GLP-1 receptor agonist GI adverse events as a clinically relevant safety signal (PMID 40499738), relevant to the rapidly expanding use of semaglutide and similar agents in T2D and obesity.
This is a shared conversation. Sign in to Orris to start your own chat.