Why anencephaly cause maternal polyhydraminos
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Here is a complete explanation based directly from the textbooks:
Why Anencephaly Causes Maternal Polyhydramnios
What is Anencephaly?
Anencephaly results from failure of the cephalic part of the neural tube to close (exencephaly). Without closure, the vault of the skull does not form, the exposed brain tissue degenerates from contact with amniotic fluid, and the brainstem is the only neural structure that remains. The cerebral cortex - including the swallowing center - is absent or non-functional.
The Core Mechanism: Absent Swallowing Reflex
Normally, the fetus actively swallows amniotic fluid throughout the second and third trimesters. This swallowing is one of the major mechanisms by which amniotic fluid volume is regulated and kept in balance.
In anencephaly:
- The cerebral cortex and higher brain centers are absent - the neural circuitry required to initiate and coordinate swallowing does not exist
- The fetus therefore cannot swallow amniotic fluid
- Fluid continues to be produced (fetal urine, lung fluid secretions) but is not being removed by swallowing
- Over the last 2 months of pregnancy, this accumulation leads to polyhydramnios (excess amniotic fluid, typically 1,500-2,000 mL or more)
Additionally, there is a secondary contributor: the exposed, ulcerated brain tissue on the surface of an anencephalic fetus transudates fluid directly into the amniotic cavity, further adding to the fluid volume.
Normal Amniotic Fluid Regulation (for context)
| Source of fluid IN | Source of fluid OUT |
|---|---|
| Fetal urine (main source, 2nd/3rd trimester) | Fetal swallowing (main route) |
| Lung fluid secretions | Intramembranous absorption |
| Oral/tracheal secretions | Transmembranous flow |
When swallowing is blocked - whether by anencephaly, esophageal atresia, or other CNS defects - the "OUT" pathway is blocked and polyhydramnios results.
Clinical Points
- Polyhydramnios from anencephaly is most prominent in the last 2 months of pregnancy
- Anencephaly can be detected on ultrasound (absent skull vault is the hallmark sign)
- Elevated maternal serum and amniotic fluid AFP (alpha-fetoprotein) is also a marker, as neural tissue is exposed to amniotic fluid
- Anencephaly occurs in ~1 per 5,000 births, is more common in females, and is always fatal
- It falls under the category of neural tube defects, most of which can be prevented by maternal folic acid supplementation (400 µg daily) before and during pregnancy
Summary in One Sentence
Anencephalic fetuses lack the cerebral swallowing centers, so they cannot swallow amniotic fluid - this impairs the primary route of fluid removal and causes progressive polyhydramnios in the third trimester.
Sources: Langman's Medical Embryology (p. 359-360, Cranial Defects); Langman's Medical Embryology (p. 139, Amniotic Fluid); Tietz Textbook of Laboratory Medicine, 7th Ed.
Spina bifida
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
I now have comprehensive textbook content and images to provide a complete answer.
Spina Bifida
Definition and Embryological Basis
Spina bifida is a neural tube defect (NTD) caused by failure of fusion of the vertebral arches (and possibly the underlying neural tube) during the 4th week of embryonic development, specifically when the caudal neuropore fails to close. This leaves the posterior elements of the spinal column - and variably the spinal cord and meninges - incompletely formed.
- More common in girls than boys
- Incidence of spina bifida cystica: ~1 in 5,000 births (considerable geographic variation)
- Strongly linked to folic acid deficiency - maternal supplementation (400 µg/day) significantly reduces incidence
- Most commonly affects the lumbar vertebrae
Classification: Four Types
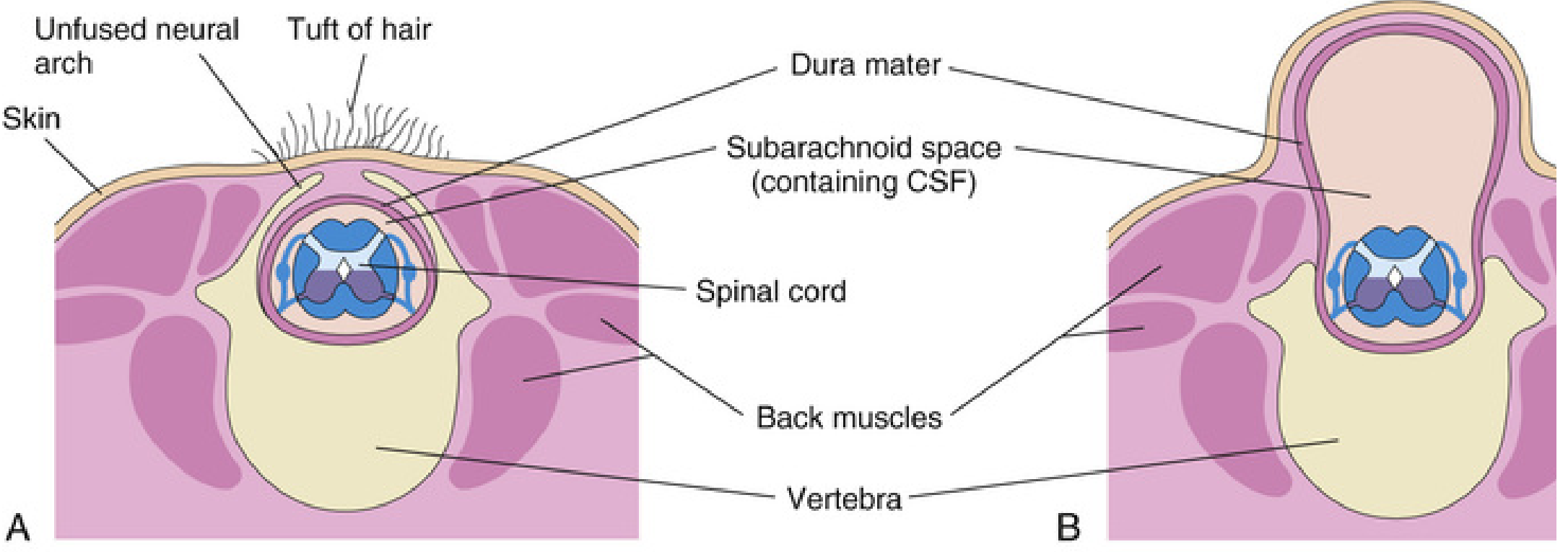
1. Spina Bifida Occulta
- Bony defect only - the neural arches of L5 or S1 fail to fuse in the midline, but there is no herniation of neural tissue
- Found in ~10% of otherwise normal people - usually an incidental finding
- May be marked externally by a small dimple, tuft of hair (hypertrichosis), lipoma, dermal sinus, or birthmark over the lumbosacral region
- Usually asymptomatic; rarely causes significant spinal cord or nerve root deficits
- The dimple marks the site where the caudal neuropore closed at the end of week 4
2. Spina Bifida with Meningocele
- Meninges (and CSF) herniate through the bony defect forming a cyst-like sac; spinal cord and nerve roots remain in their normal position
- The sac is covered by skin
- Neurological deficits may be absent or mild
- If skin cover is adequate, may be managed conservatively; a large sac can be surgically excised at 3-6 months
3. Spina Bifida with Meningomyelocele (the most common severe form)
- Both meninges and spinal cord/nerve roots herniate into the sac
- The roof of the defect is formed by exposed neural tissue (not covered by normal skin)
- Causes motor, sensory, and autonomic deficits below the level of the lesion
- Dermatomal loss of sensation, skeletal muscle paralysis, and sphincter dysfunction (bladder/bowel) are common
- 75-90% of cases develop hydrocephalus, nearly always due to the associated Arnold-Chiari type II malformation
- Most lesions are lumbosacral
4. Spina Bifida with Myeloschisis (Rachischisis)
- The most severe form - the neural tube lies completely open; the spinal cord is a flat, exposed plate of neural tissue
- No cyst; the cord is directly exposed to amniotic fluid
- Severe neurological deficits; poor prognosis
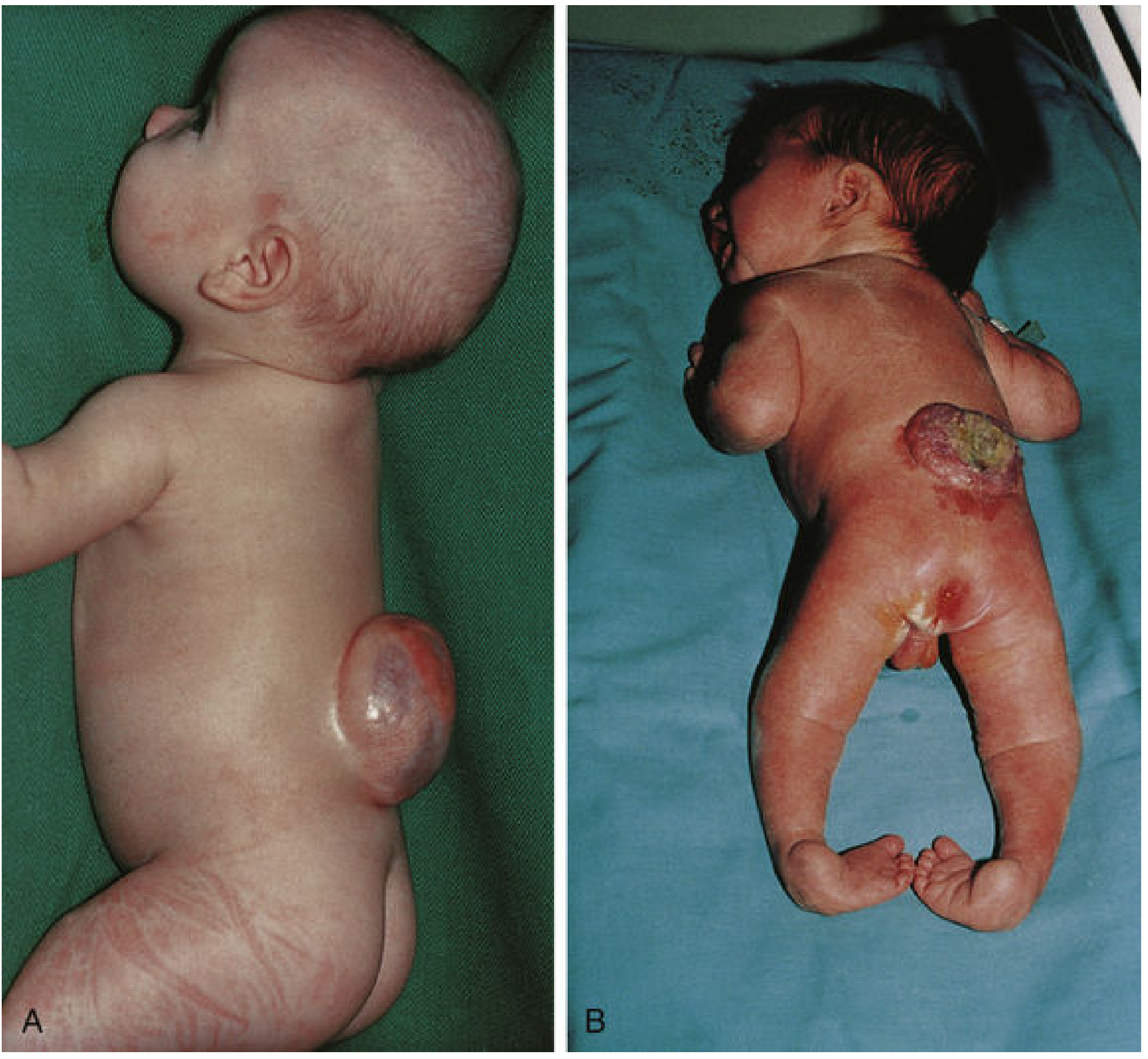
Fig. A: Spina bifida with meningomyelocele (smooth covered sac). Fig. B: Spina bifida with myeloschisis - open exposed cord with affected lower limbs.
Types Summary Table
| Type | Bony defect | Meninges | Spinal cord/roots | Skin cover | Neurological deficit |
|---|---|---|---|---|---|
| Occulta | Yes | No herniation | Normal | Intact | Usually none |
| Meningocele | Yes | Herniated (CSF) | Normal position | Thin/intact | Mild or none |
| Meningomyelocele | Yes | Herniated | Herniated into sac | Absent/incomplete | Moderate-severe |
| Myeloschisis | Yes | Open | Exposed plate | Absent | Severe |
Associated Abnormalities
Arnold-Chiari Type II Malformation
- Present in almost all infants with spina bifida (myelomeningocele)
- The medulla oblongata and cerebellar tonsils herniate downward through the foramen magnum into the cervical spinal canal
- Causes pressure on the lower medulla
- Strongly associated with obstructive hydrocephalus (blockage of CSF flow)
- Features: headache, vomiting, visual disturbances, cerebellar ataxia, sensory disturbances, or paralysis
Hydrocephalus
- Occurs in 75% of meningomyelocele cases
- Mostly due to the Arnold-Chiari malformation blocking CSF outflow
- Requires surgical CSF diversion (ventriculoperitoneal shunt)
Tethered Cord Syndrome
- Progressive neurological deficit due to spinal cord tethering and traction as the child grows
- Part of the broader group of spinal dysraphism disorders
Neuropathic Bladder
- Common long-term complication of lumbosacral meningomyelocele
- Leads to urinary incontinence, recurrent UTIs, and upper tract damage
Neurological Deficits by Level
| Level | Motor deficit | Sensory loss | Bladder/bowel |
|---|---|---|---|
| Thoracic | Paraplegia | Trunk/legs | Yes |
| Lumbar (L1-L3) | Hip flexion/adduction weakness | Thigh/leg | Yes |
| Lumbar (L4-L5) | Foot drop, knee instability | Below knee | Common |
| Sacral | Foot/ankle weakness | Perianal (saddle) | Common - sphincter paralysis |
Prenatal Diagnosis
- Ultrasound: absence of posterior vertebral arches; banana sign (cerebellum pulled caudally) and lemon sign (frontal bone scalloping) due to associated Chiari malformation
- Elevated maternal serum AFP: leaked from exposed neural tissue into amniotic fluid and then maternal blood. Used in routine antenatal screening (15-20 weeks)
- Amniotic fluid AFP and acetylcholinesterase: confirmatory if serum AFP elevated
Management
Immediate (Neonatal)
- Cover the defect with sterile moist dressings to protect neural tissue
- Early surgical closure within 24-48 hours to prevent meningitis and CSF contamination
- Treat hydrocephalus with ventriculoperitoneal (VP) shunt
Selective Approach for Myelomeningocele
- Previously, all defects were closed surgically; now a selective approach is used
- Surgery is considered inappropriate in infants with severe hydrocephalus, large open defect, and no distal neurological function - majority of these infants die in the first year without closure
- With antibiotics, early surgical closure, and shunting: ~50% of children who survive the first 24 hours reach school age
Fetal Surgery
- In utero repair of myelomeningocele (before 26 weeks gestation) reduces the need for shunting and improves motor outcomes (MOMS trial)
Long-term Multidisciplinary Care
- Neuropathic bladder - clean intermittent catheterization (CIC), urodynamics
- Neuromuscular scoliosis - bracing or surgical correction
- Bone and joint deformity
- Skin care (pressure sores due to sensory loss)
- Cognitive and developmental support
Prevention
Folic acid 400 µg/day before conception and during the first trimester significantly reduces the risk of neural tube defects. Fortification of diets (bread/cereals) with folic acid has led to a declining incidence of spina bifida in many countries.
Sources: The Developing Human - Clinically Oriented Embryology (p. 1040-1046); Bailey & Love's Short Practice of Surgery 28th Ed. (p. 544, 684); Bradley & Daroff's Neurology in Clinical Practice; Langman's Medical Embryology; Campbell-Walsh-Wein Urology; The Developing Human (p. 928)
Why and how meningomyelocele causes hydrocepwhuksu
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have everything needed for a complete, well-illustrated answer.
How Myelomeningocele Causes Hydrocephalus
The answer has two layers: why the Chiari II malformation develops (the root cause), and how it then blocks CSF flow to produce hydrocephalus.
Step 1 - The Root Cause: Why Does Myelomeningocele Lead to Chiari II?
The most widely accepted explanation is the Unified Theory:
"Failure of closure of the neural tube results in CSF leakage through the open spinal defect, leading to lack of development of the posterior fossa and abnormalities of neural and calvarial development."
- Creasy & Resnik's Maternal-Fetal Medicine
Here is the sequence:
- The neural tube fails to close in the lumbosacral region at ~week 4 of gestation
- The open myelomeningocele defect allows CSF to leak freely out of the spinal canal into the amniotic fluid throughout fetal development
- Because CSF is continuously lost, there is insufficient hydraulic pressure inside the developing neural tube
- This low pressure means the posterior fossa (the bony space at the back of the skull housing the brainstem and cerebellum) does not expand properly - it remains abnormally small
- The cerebellar vermis, tonsils, fourth ventricle, and medulla are forced downward through the enlarged foramen magnum into the cervical spinal canal - this is the Arnold-Chiari type II malformation
In short: CSF leak → small posterior fossa → brain herniates downward through foramen magnum.
Step 2 - How Chiari II Produces Hydrocephalus
The herniated posterior fossa structures mechanically obstruct CSF circulation at two key points:
Obstruction at the Foramen Magnum
- The herniated medulla, cerebellar vermis and tonsils physically plug the foramen magnum
- CSF flowing down from the ventricular system cannot pass freely around the craniocervical junction
- This creates a non-communicating (obstructive) hydrocephalus
Obstruction of the Fourth Ventricle
- The fourth ventricle is dragged downward and becomes elongated and slit-like
- Its outlets (foramina of Luschka and Magendie) are displaced and compressed
- CSF cannot exit the fourth ventricle normally
Aqueductal Stenosis (secondary)
- The midbrain shows "beaking" of the tectum (the posterior midbrain appears elongated on sagittal MRI)
- This distortion can narrow the cerebral aqueduct (of Sylvius), adding another level of obstruction between the third and fourth ventricles
The result: CSF produced in the lateral ventricles by the choroid plexus cannot drain through its normal pathway → all ventricles proximal to the obstruction dilate → hydrocephalus.
The Full Pathway (Summary)
Myelomeningocele (open neural tube defect)
↓
CSF leaks out of spinal canal throughout fetal life
↓
Posterior fossa remains small / underdeveloped
↓
Cerebellum + brainstem + 4th ventricle herniate
downward through foramen magnum
↓
= Arnold-Chiari Type II Malformation
↓
Obstructs CSF outflow at:
• Foramen magnum (physical plug)
• 4th ventricle outlets (Luschka, Magendie)
• Cerebral aqueduct (beaking/kinking)
↓
Non-communicating (obstructive) hydrocephalus
(entire ventricular system dilates)
Key Numbers
| Fact | Figure |
|---|---|
| Myelomeningocele cases with Chiari II | ~100% |
| Myelomeningocele cases developing hydrocephalus | 80-90% |
| Hydrocephalus present at birth | ~25% |
| Hydrocephalus developing after postnatal MMC repair | ~80% of remaining cases |
| Cases with symptomatic brainstem dysfunction | ~20% (but high mortality) |
Why does hydrocephalus often only appear after surgical repair of the MMC? Before repair, the open defect acts as a "pressure release valve" - CSF is still leaking out. Once the defect is surgically closed, CSF can no longer escape, the obstruction at the foramen magnum becomes fully effective, and hydrocephalus rapidly develops.
MRI Appearances of Chiari II
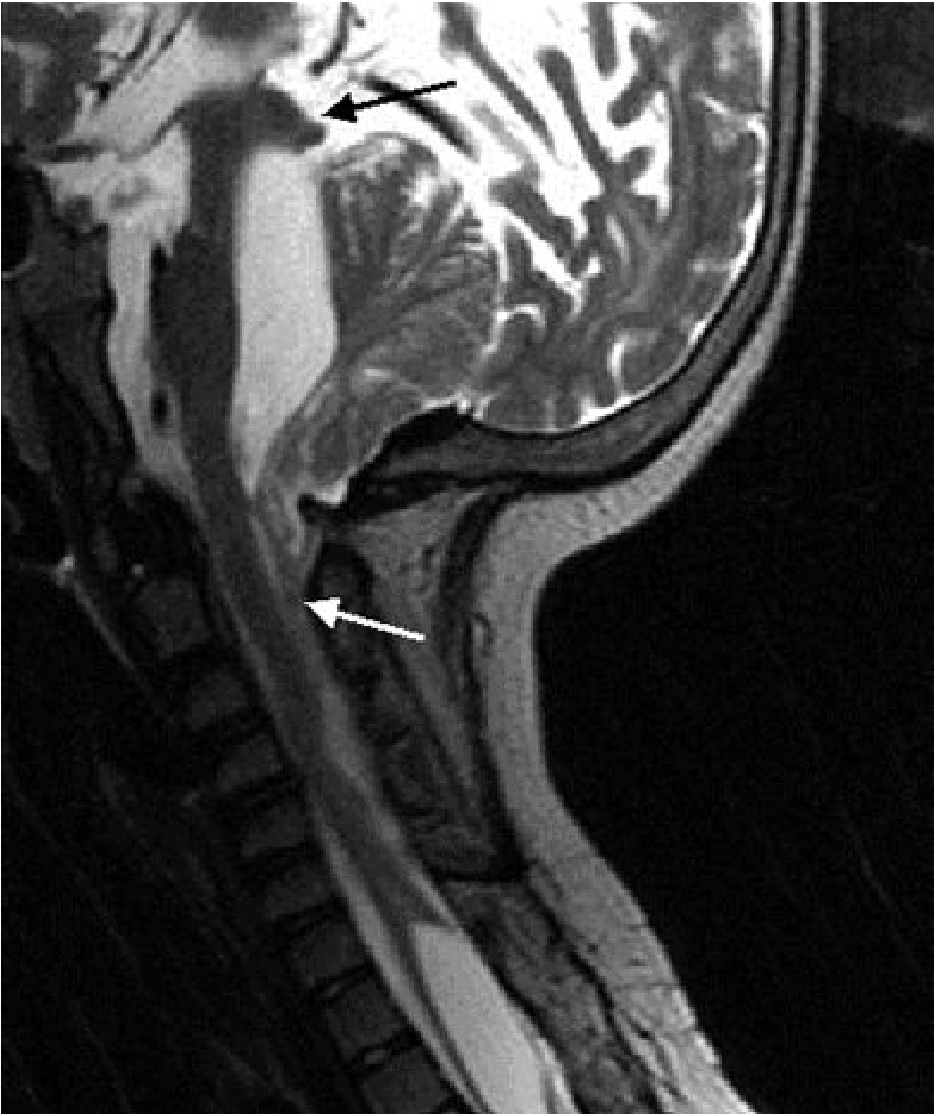
Sagittal MRI of Chiari II: the cerebellum herniates superiorly through the tentorium (black arrow) and the brainstem/cord descend into the cervical canal (white arrow). The posterior fossa is small.
Key MRI signs:
- Small, shield-shaped posterior fossa
- Downward displacement of cerebellar vermis, tonsils, 4th ventricle, medulla
- "Beaking" of the midbrain tectum
- Slit-like fourth ventricle (normal/enlarged = suggests isolated 4th ventricle or hydrocephalus)
- Medullary kinking
- Enlarged massa intermedia
- On antenatal ultrasound: "banana sign" (compressed, curved cerebellum) and "lemon sign" (frontal bone scalloping due to caudal traction on the brain)
Treatment of the Hydrocephalus
- Ventriculoperitoneal (VP) shunt - traditional standard; diverts CSF from the lateral ventricles to the peritoneal cavity
- Endoscopic Third Ventriculostomy (ETV) + Choroid Plexus Cauterization (CPC) - avoids a permanent shunt; effective in >70% of MMC patients
- Fetal surgery (in utero MMC repair before 26 weeks) - reduces the need for shunting by allowing the posterior fossa to develop more normally before birth; 40% still require shunting despite prenatal repair (MOMS trial data)
- Posterior fossa decompression - for symptomatic brainstem compression from the Chiari herniation itself
Sources: Creasy & Resnik's Maternal-Fetal Medicine (Chiari II - Unified Theory); The Developing Human - Clinically Oriented Embryology (p. 1083); Grainger & Allison's Diagnostic Radiology (p. 1976-1977); Goldman-Cecil Medicine; Campbell-Walsh-Wein Urology; Barash Clinical Anesthesia
Why there is dilated 4 ventricle with absent cerebellum in dandy walker malformation
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I have all the information needed. Here is the complete answer.
Dandy-Walker Malformation: Why the 4th Ventricle is Dilated and the Cerebellum is Absent
What is Dandy-Walker Malformation?
Dandy-Walker malformation (DWM) is a congenital posterior fossa malformation characterized by three cardinal features:
- Cystic dilatation of the 4th ventricle (the 4th ventricle balloons into a large posterior fossa cyst)
- Aplasia or hypoplasia of the cerebellar vermis (the midline portion of the cerebellum is absent or rudimentary)
- Enlarged posterior fossa with upward displacement of the tentorium, torcula, and straight sinus above the lambdoid suture
The Core Developmental Problem
The key is understanding what normally happens during embryonic development of the 4th ventricle roof.
Normal Development
The roof of the 4th ventricle is initially a thin membrane called the posterior membranous area (Blake's pouch). During normal development:
- This membrane perforates at around week 26 of gestation to form the foramen of Magendie (median aperture) in the midline
- The foramina of Luschka (lateral apertures) also open
- These three openings allow CSF produced inside the 4th ventricle to exit freely into the subarachnoid space
- With free CSF egress, the 4th ventricle remains normally sized
- Simultaneously, CSF pressure inside the developing posterior fossa drives normal expansion of the cerebellar vermis and hemispheres
What Goes Wrong in Dandy-Walker
The foramina of Luschka and Magendie fail to perforate/develop properly.
The sequence that follows:
Foramina of Luschka & Magendie fail to open
↓
CSF cannot exit the 4th ventricle
↓
CSF accumulates inside the 4th ventricle
↓
4th ventricle progressively expands into a large cyst
(= "roofless 4th ventricle" lined by ependyma)
↓
The ballooning cyst DISPLACES and COMPRESSES the
developing cerebellar vermis rather than allowing
it to grow → vermis is absent or hypoplastic
↓
Posterior fossa enlarges to accommodate the cyst
Tentorium and torcula are pushed upward
↓
Hydrocephalus (obstructive) from blocked CSF outflow
"This cyst represents the expanded, roofless fourth ventricle in the absence of a normally formed vermis."
- Robbins & Kumar Pathologic Basis of Disease
Why Specifically is the Vermis Absent?
Two concurrent mechanisms:
1. Primary Developmental Failure
The cerebellar vermis (midline cerebellum) simply fails to develop normally - there is a primary defect in midline cerebellar formation. This is the underlying embryological insult. The lateral cerebellar hemispheres may be relatively preserved or also hypoplastic.
2. Mechanical Compression/Displacement by the Expanding Cyst
The progressively enlarging 4th ventricle cyst physically prevents the vermis from forming and occupies the space the vermis should inhabit. The thin rim of cerebellar tissue seen on imaging forms the lateral wall of the posterior fossa cyst.
The 4th ventricle in DWM is described as "open" at the back - it has no posterior wall formed by the vermis (which is absent). Instead, its posterior surface communicates directly with the cyst which fills the enlarged posterior fossa.
The MRI - What You See
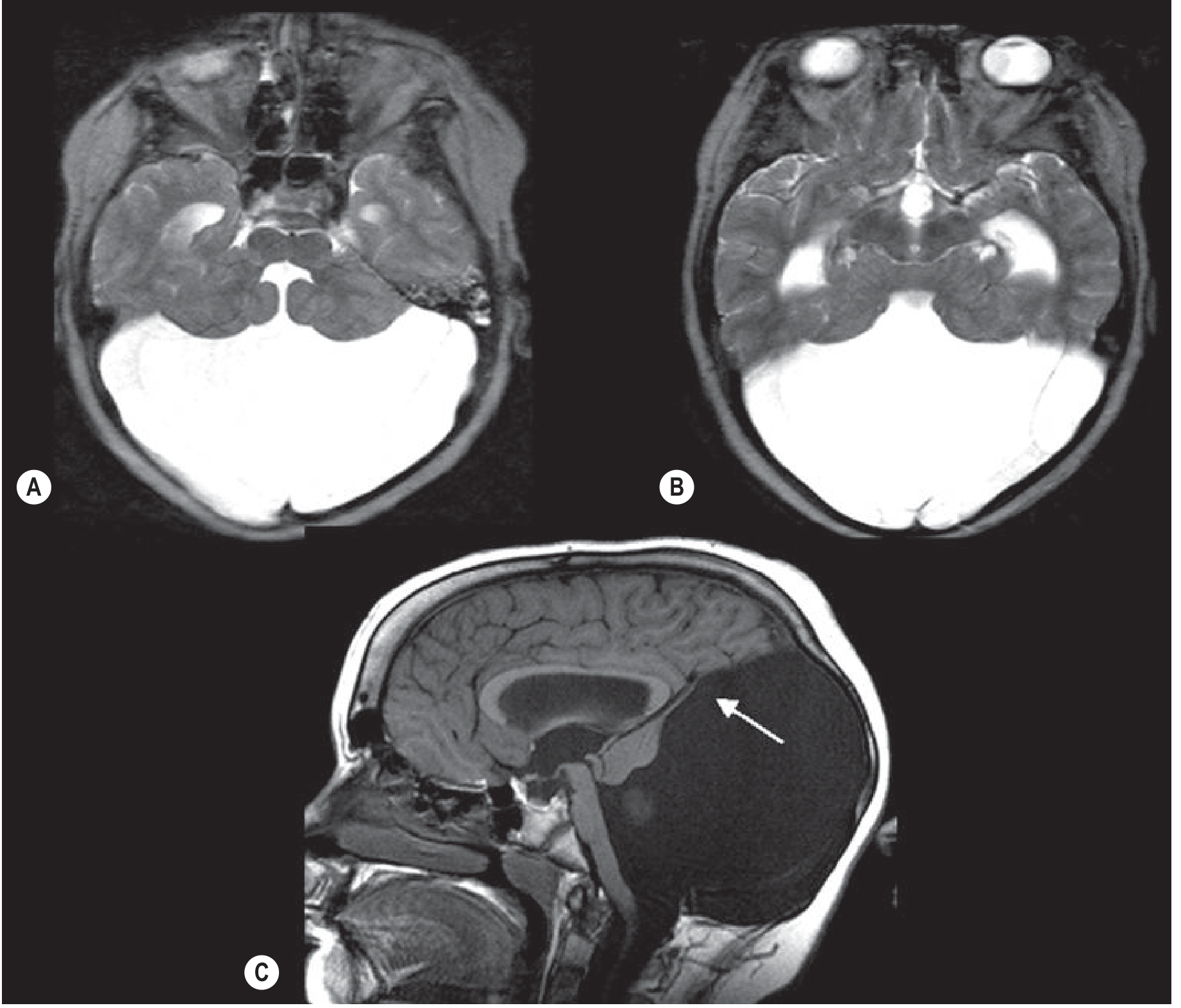
Fig. 76.7 (Grainger & Allison): (A & B) The 4th ventricle opens into a large posterior fossa cyst with hydrocephalus. (C) Hypoplastic cerebellum - only a thin rim of cerebellar tissue (arrow) forms the cyst wall. The venous confluence and tentorium are elevated above the lambdoid suture.
Key imaging findings:
| Feature | Explanation |
|---|---|
| Large posterior fossa cyst | Expanded, roofless 4th ventricle |
| Absent/hypoplastic cerebellar vermis | Primary failure of vermis formation + compression |
| Elevated tentorium/torcula above lambdoid suture | Posterior fossa expansion pushes tentorium up |
| Hydrocephalus (dilated lateral and 3rd ventricles) | Obstruction of CSF outflow from 4th ventricle |
| Thin rim of cerebellar tissue at cyst wall | Compressed, displaced cerebellar remnant |
| Dilated cerebral aqueduct + 3rd ventricle | Retrograde obstruction from blocked 4th ventricle outflow |
Associated Features and Anomalies
- Corpus callosum agenesis/hypoplasia - in ~20-25% of cases
- Heterotopia of inferior olivary nuclei
- Pachygyria of the cerebral cortex
- Hydrocephalus - develops in the majority due to obstruction; may not be present at birth but emerges in infancy
- Brainstem nuclei dysplasias - often found on pathology
- Other midline anomalies (septal, hypothalamic)
- Can be associated with chromosomal abnormalities and genetic syndromes
Dandy-Walker Spectrum (from severe to mild)
| Variant | 4th Ventricle | Vermis | Posterior Fossa | Hydrocephalus |
|---|---|---|---|---|
| Classic DWM | Massively dilated cyst | Absent/aplastic | Greatly enlarged | Common |
| DW Variant | Mildly enlarged | Hypoplastic (partial) | Mildly enlarged | Variable |
| Blake's Pouch Cyst | Mildly enlarged | Normal | Normal/mildly enlarged | Possible |
| Mega Cisterna Magna | Normal | Normal | Enlarged cisterna only | Absent |
Blake's Pouch Cyst is an important variant: the posterior membranous area (Blake's pouch) fails to perforate at the normal time (week 26), so the cyst remains attached to the 4th ventricle roof. Unlike classic DWM, the vermis and cerebellar hemispheres develop normally because the anterior membranous area is intact.
Contrast with Chiari II
| Dandy-Walker | Chiari II | |
|---|---|---|
| 4th ventricle | Massively dilated/cystic | Small, slit-like, displaced downward |
| Cerebellum | Vermis absent, cyst fills posterior fossa | Herniated downward through foramen magnum |
| Posterior fossa | Enlarged | Small |
| Tentorium | Elevated | Low |
| Association | Isolated or genetic syndromes | Myelomeningocele (nearly 100%) |
| Mechanism of hydrocephalus | Blocked 4th ventricle outlets (foramina) | Foramen magnum obstruction by herniated tissue |
Clinical Presentation
- Macrocephaly (enlarging head circumference) - often the first sign in infancy
- Bulging posterior fontanelle
- Signs of raised intracranial pressure (irritability, vomiting, sunset sign)
- Developmental delay - more related to associated brain anomalies than hydrocephalus alone
- Cerebellar signs - ataxia, hypotonia (if cerebellar tissue remains)
- Seizures - from associated cortical dysplasias
Treatment
- VP shunt for hydrocephalus - drains the cyst directly (cystoperitoneal shunt) or lateral ventricles
- Management of associated anomalies
- Prognosis depends heavily on associated brain malformations, not just the posterior fossa cyst
Sources: Bradley & Daroff's Neurology in Clinical Practice (p. 1884, 2555); Grainger & Allison's Diagnostic Radiology (p. 1973); Robbins & Kumar Pathologic Basis of Disease; Adams & Victor's Principles of Neurology
Why these Forman fail to perforate
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Searching the Web
Dandy-Walker malformation why foramina Luschka Magendie fail to open causes genetics pathogenesis
Reading Web Page
https://en.wikipedia.org/wiki/Dandy%E2%80%93Walker_malformat…
I now have everything needed to give a thorough, honest, and up-to-date answer - including the important caveat that the classical teaching is outdated.
Why Do the Foramina of Luschka and Magendie Fail to Perforate in Dandy-Walker?
The Honest Answer First
This is a question where the classical teaching and the modern understanding disagree, and it is important to know both.
Classical teaching (Taggart & Walker, 1942): Dandy-Walker is caused by atresia (failure to open) of the foramina of Luschka and Magendie, trapping CSF in the 4th ventricle.
Modern view: This explanation is no longer believed to be fully correct. Later studies showed that in most DWM patients, these foramina are actually open (or become open). The pathogenesis is now understood to be a primary developmental failure of the cerebellar vermis and posterior fossa mesenchyme, not simply foraminal atresia. The foraminal obstruction, when present, is a contributing factor rather than the root cause.
"Taggart and Walker... ascribed the potential cause as underdevelopment of the foramina of Luschka and Magendie - now no longer believed to be significant." - Wikipedia (citing updated literature)
What Actually Causes the Developmental Arrest?
The underlying event is a developmental arrest in the hindbrain occurring between weeks 4 and 6 of gestation (Creasy & Resnik). The exact trigger is multifactorial and in most cases unknown, but the known causes are:
1. Genetic Causes (most important)
| Gene/Chromosome | Mechanism |
|---|---|
| ZIC1 and ZIC4 (chromosome 3q) | Zinc finger transcription factors critical for cerebellar vermis development; deletion causes DWM |
| ZIC2 and ZIC5 (chromosome 13q) | Involved in Blake's pouch cyst variant (haploinsufficiency from 13q deletion) |
| FOXC1 | Forkhead transcription factor; mutations linked to DWM, particularly involving mesenchymal development of the posterior fossa |
| Trisomy 18 (most common trisomy association) | Extra chromosome 18 - DWM is the most frequent trisomy association |
| Trisomy 13, 21, 9 | Also associated with DWM |
| Chromosomal deletions/duplications | Various partial chromosomal imbalances |
| Autosomal recessive/dominant inheritance | Rare familial forms exist |
The key pathway: these genes regulate mesenchymal development of the posterior fossa and rhombic lip development (the germinal zone that gives rise to the cerebellar vermis). When they fail, the vermis does not form properly AND the posterior fossa mesenchyme does not develop correctly, meaning the 4th ventricle roof membrane never gets the proper developmental signal to perforate.
2. The Blake's Pouch Theory (Developmental Timing Failure)
This is the best current embryological explanation for why the foramen fails to open:
- The roof of the 4th ventricle passes through a stage called Blake's Pouch - a blind-ended invagination
- Normally at ~week 26 of gestation, this pouch ruptures/perforates to form the foramen of Magendie
- If the posterior membranous area fails to regress at the programmed time, the pouch remains intact as a cyst
- In classic DWM, the primary failure is of cerebellar vermis formation (not foraminal atresia) - the vermis simply never develops, so the posterior wall of the 4th ventricle is never properly formed by neural tissue
- In Blake's Pouch Cyst (a milder variant), the vermis IS normal but the membrane just fails to perforate - this is the purer "foraminal failure" form
3. Environmental / Teratogenic Causes
These disrupt normal hindbrain/mesenchymal development during weeks 4-6:
| Teratogen | Evidence |
|---|---|
| Warfarin (during pregnancy) | Linked to DWM in 1985; also causes corpus callosum agenesis and ocular dysgenesis |
| Maternal diabetes | Increased risk of DWM |
| Alcohol | Suggested but uncertain evidence |
| Viral infections (congenital) | Cytomegalovirus, rubella - disrupt hindbrain development |
| Isotretinoin (vitamin A derivative) | Hindbrain teratogen |
The Underlying Mechanism - Putting It Together
Primary defect: Genetic mutation OR teratogen
(weeks 4-6 of gestation)
↓
Abnormal development of:
• Rhombic lip (germinal zone → cerebellar vermis)
• Posterior fossa mesenchyme
• Posterior membranous area (Blake's pouch) timing
↓
Cerebellar vermis fails to form (aplasia/hypoplasia)
↓
The posterior wall of the 4th ventricle is never
properly "closed" by vermian tissue
↓
Blake's pouch / posterior membranous area fails to
perforate (secondary to the primary malformation)
↓
4th ventricle communicates with an expanding
posterior fossa cyst instead of the subarachnoid space
↓
Progressive cyst enlargement → hydrocephalus
(in ~70-80% of cases)
Key Point: Foraminal Atresia vs. Primary Vermian Failure
| Classical teaching | Modern understanding |
|---|---|
| Foramina fail to open → CSF trapped → 4th ventricle dilates → compresses vermis | Primary failure of vermis/posterior fossa development → 4th ventricle has no posterior wall → acts as an open cyst |
| Foraminal atresia is the PRIMARY cause | Foraminal non-patency is SECONDARY to primary mesenchymal/cerebellar maldevelopment |
| Analogous to a "blocked drain" | More like a "drain that was never properly built" |
Why is the Cause Frequently Unknown?
In most sporadic (non-familial) isolated DWM cases, no specific cause is identified. This is because:
- Many responsible gene mutations have not yet been discovered
- The condition is genetically heterogeneous (many different genes can produce the same phenotype)
- Environmental contributions are hard to confirm retrospectively
- The event occurs very early (weeks 4-6), often before a woman knows she is pregnant
Sources: Bradley & Daroff's Neurology in Clinical Practice (p. 1884); Creasy & Resnik's Maternal-Fetal Medicine (p. 395); MedlinePlus Genetics - Dandy-Walker Malformation (FOXC1, ZIC1, ZIC4); Wikipedia - Dandy-Walker Malformation (updated pathophysiology); ISUOG Fetal Brain Imaging
AC malformation type ll how occur in ass with syringomyelia
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Excellent - I now have rich, detailed textbook content from Adams & Victor covering all the major theories. I have everything I need.
Arnold-Chiari Type II and Syringomyelia: How They Are Connected
What is a Syrinx?
A syrinx (syringomyelia) is a fluid-filled cavity within the spinal cord parenchyma, usually in the central/paracentral cervical region. It is not simply a dilated central canal (which is called hydromyelia) - it is a pathological cavity that can extend up into the brainstem (syringobulbia) or down into the thoracic cord.
The key question is: how does the Chiari II herniation at the foramen magnum create a cavity deep inside the spinal cord?
The Fundamental Problem: Blocked CSF Flow at the Foramen Magnum
In Arnold-Chiari type II, the cerebellar vermis, tonsils, brainstem, and 4th ventricle are all displaced downward through the foramen magnum into the cervical spinal canal. This herniated tissue:
- Partially or completely occludes the subarachnoid space at the craniocervical junction
- Disrupts the normal two-way pulsatile flow of CSF between the cranial and spinal compartments
Under normal conditions, with every heartbeat:
- Systolic arterial pulsation causes a brief rise in intracranial pressure
- CSF surges smoothly downward through the foramen magnum into the spinal subarachnoid space
- It then returns during diastole
- This pulsatile flow is free and symmetric
When the Chiari herniation plugs the foramen magnum, this smooth flow is blocked - and it is the resulting abnormal pressure dynamics that drive fluid into the cord.
The Three Theories of How the Syrinx Forms
Theory 1 - Gardner's Hydrodynamic Theory ("Water Hammer" / Piston Theory)
(The classical explanation - still widely taught)
- The herniated tonsils block the foramina of Luschka and Magendie (or the foramen magnum)
- CSF cannot exit the 4th ventricle normally
- With each systolic pulse of the choroid plexus, a pressure wave ("water hammer effect") is transmitted down the central canal of the spinal cord
- This repeated pulsatile pressure progressively dilates and then ruptures the central canal wall, forming a syrinx
- The syrinx is therefore essentially a dilated, diverticulated central canal
"A pulse wave of CSF pressure generated by systolic pulsations of the choroid plexuses is transmitted into the cord from the fourth ventricle through the central canal... the syrinx consists essentially of a greatly dilated central canal with a diverticulum that ramifies from the central canal."
- Adams & Victor's Principles of Neurology
Limitation: This theory is now questioned because: (a) in many cases the foramina are actually patent; (b) no direct connection between the 4th ventricle and the syrinx can be found on histology; (c) the calculated pressure wave amplitude is probably too small.
Theory 2 - Oldfield/Heiss "Piston Effect" / Subarachnoid Pressure Theory
(Currently the most accepted modern theory)
- The herniated cerebellar tonsils act as a piston in a cylinder (the spinal canal)
- With each heartbeat, the tonsils are pushed slightly downward by the intracranial pulsation
- This compresses the spinal subarachnoid CSF from outside the cord
- The pressure wave drives CSF into the spinal cord parenchyma through Virchow-Robin perivascular spaces and other subpial channels (NOT through the central canal)
- Over time, these small fluid collections coalesce to form a syrinx cavity
- The syrinx initially forms outside the central canal, but eventually connects to it (hydromyelia ex vacuo)
"The compressive effect of the cerebellar tonsils partially occludes the subarachnoid space at the foramen magnum and creates pressure waves that compress the spinal cord from without and not from within; the pressure waves propagate syrinx fluid caudally with each heartbeat."
- Adams & Victor's Principles of Neurology (Heiss et al.)
This explains why:
- The syrinx can form without a direct 4th ventricle-to-central canal connection
- Decompression of the foramen magnum (removing the "piston") often causes the syrinx to shrink or disappear
Theory 3 - Virchow-Robin Perivascular Tracking
(Related to Theory 2)
- Subarachnoid obstruction at the craniocervical junction increases spinal CSF pressure during straining, coughing, or physical effort
- Under this elevated pressure, CSF tracks inward along Virchow-Robin (perivascular) spaces into the cord parenchyma
- Small fluid pools accumulate and coalesce over years into a syrinx cavity
- This also explains why coughing or straining worsens symptoms acutely in syringomyelia
Why Chiari II Specifically (vs. Chiari I)?
| Chiari I | Chiari II | |
|---|---|---|
| Structures herniated | Cerebellar tonsils only | Tonsils + vermis + 4th ventricle + medulla |
| Foramen magnum occlusion | Partial | More complete |
| Association with syrinx | Common (majority of syringomyelia cases) | Present but often overshadowed by hydrocephalus |
| Timing of symptoms | Adolescence/adulthood | Infancy (due to MMC/hydrocephalus) |
Chiari I is actually the more common cause of syringomyelia in clinical practice. In Chiari II, the syrinx may be present but is often less prominent because hydrocephalus is the dominant problem. Treating the hydrocephalus (with a VP shunt) often reduces the syrinx in Chiari II.
The Full Sequence - Visual Summary
Arnold-Chiari II Malformation
(herniated tonsils, vermis, medulla through foramen magnum)
↓
Foramen magnum partially/fully occluded
↓
BLOCKED pulsatile CSF flow at craniocervical junction
↓
With each heartbeat...
↓
┌─────────────────────────────────┐
│ Theory 1 (Gardner) │ Theory 2 (Heiss/Oldfield)
│ Pressure wave travels DOWN │ Tonsils act as PISTON
│ central canal of cord │ Compress cord from OUTSIDE
│ → dilates central canal │ → CSF enters via
│ → syrinx (dilated canal) │ Virchow-Robin spaces
└─────────────────────────────────┘
↓
Progressive fluid accumulation inside cord parenchyma
↓
SYRINX (fluid-filled cavity, usually cervical cord)
↓
May extend upward into brainstem = SYRINGOBULBIA
Clinical Features of Syringomyelia (when it complicates Chiari II)
The syrinx destroys the central cord where the pain and temperature fibers cross (anterior commissure), while initially sparing the peripherally located touch, vibration, and proprioception fibers:
| Symptom | Mechanism |
|---|---|
| Dissociated sensory loss - loss of pain & temperature, preserved touch/vibration | Central cord destruction at anterior commissure (crossing spinothalamic fibers) |
| Cape/shawl distribution of sensory loss (neck, shoulders, arms) | Cervical syrinx |
| Painless injuries and burns of the hands | Loss of pain sensation |
| Hand/arm weakness and wasting (lower motor neuron) | Anterior horn cell destruction |
| Absent upper limb reflexes | Arc disrupted at cervical level |
| Spastic paraplegia (if large syrinx) | Corticospinal tract involvement |
| Scoliosis | Asymmetric muscle weakness |
| Syringobulbia (if extends upward) | Nystagmus, tongue weakness, dysphagia, facial sensory loss |
Treatment Implication
The key insight from the Heiss/Oldfield theory is that decompression of the foramen magnum removes the "piston effect":
- Posterior fossa decompression (craniectomy + C1 laminectomy ± duraplasty) - restores normal CSF flow at the craniocervical junction
- The syrinx often collapses or shrinks after decompression without directly treating the syrinx
- In Chiari II with hydrocephalus, VP shunting reduces intracranial pressure and can indirectly resolve the syrinx
- Direct syrinx shunting (syringoperitoneal shunt) is reserved for cases that fail decompression
Sources: Adams & Victor's Principles of Neurology, 12th Ed. (p. 1278-1282, Syringomyelia - Pathogenesis of Type I); Morgan & Mikhail's Clinical Anesthesiology 7e; Bailey & Love's Short Practice of Surgery 28th Ed.; Miller's Review of Orthopaedics
Can you simplify me the spinal tract and their damage lead to which disorder
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have all the content I need, including the excellent Harrison's spinal cord cross-section diagram. Here is the complete, simplified answer.
Spinal Tracts and What Damage to Each Causes
The Spinal Cord Cross-Section - A Map
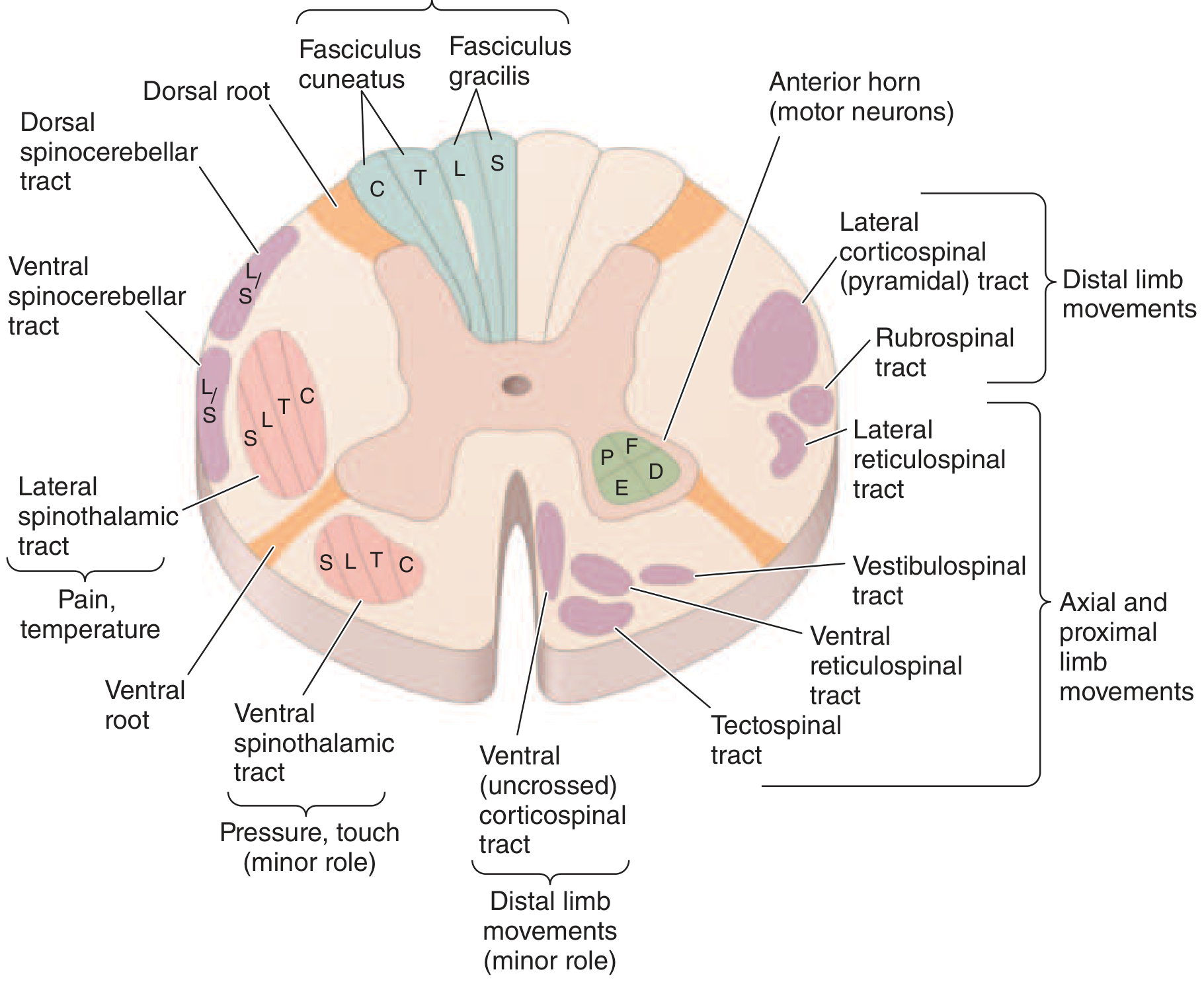
Harrison's Principles of Internal Medicine - Spinal cord cross-section showing principal tracts
Think of the spinal cord in three regions:
- Posterior (dorsal) = sensory (fine sensation)
- Lateral = motor (corticospinal) + pain/temperature (spinothalamic)
- Anterior (ventral) = motor neurons + crude touch
THE ASCENDING (SENSORY) TRACTS
1. Posterior Columns (Dorsal Columns)
Tracts: Fasciculus gracilis (legs/lower trunk) + Fasciculus cuneatus (arms/upper trunk)
| What they carry | Where they cross | Side of cord |
|---|---|---|
| Vibration, proprioception (joint position), fine touch, pressure | Cross in the medulla (high up) | Travel ipsilateral (same side) in the cord |
Damage →
- Ipsilateral loss of vibration sense and joint position below the lesion
- Sensory ataxia - patient cannot feel where their limbs are → wide-based unsteady gait
- Positive Romberg sign (falls when eyes closed - relies on vision to compensate)
- Classic diseases: Subacute combined degeneration (B12 deficiency), tabes dorsalis (syphilis), Friedreich's ataxia, posterior cord syndrome
2. Lateral Spinothalamic Tract
Tract: Crosses within 1-2 segments of entry, then ascends contralateral
| What it carries | Where it crosses | Side of cord |
|---|---|---|
| Pain and temperature | Crosses at the anterior commissure (within spinal cord) | Travels contralateral (opposite side) |
Damage →
- Contralateral loss of pain and temperature 1-2 levels below the lesion
- Touch is preserved (carried by both posterior column and ventral spinothalamic)
- Classic scenario: Syringomyelia destroys the fibers as they cross → "cape/shawl" distribution loss
- Somatotopy: sacral fibers are outermost → deep lesions spare sacral ("sacral sparing")
3. Ventral (Anterior) Spinothalamic Tract
| What it carries | Side |
|---|---|
| Crude touch and pressure (minor role) | Contralateral |
Rarely tested in isolation - light touch is mostly preserved even with damage because it has redundant pathways.
4. Spinocerebellar Tracts (Dorsal + Ventral)
| What they carry | Function |
|---|---|
| Unconscious proprioception from muscles/joints | Coordination and balance via cerebellum |
Damage →
- Ipsilateral limb ataxia (clumsy, uncoordinated movements)
- Loss of smooth, coordinated gait
- Seen in: Friedreich's ataxia, multiple sclerosis
THE DESCENDING (MOTOR) TRACTS
5. Lateral Corticospinal Tract (Pyramidal Tract) - THE MOST IMPORTANT
| Origin | Crosses | Side in cord |
|---|---|---|
| Motor cortex | At the pyramidal decussation in the medulla | Travels ipsilateral (already crossed) |
Damage → Upper Motor Neuron (UMN) syndrome:
- Ipsilateral spastic weakness below the lesion
- Increased tone (spasticity)
- Hyperreflexia (exaggerated deep tendon reflexes)
- Positive Babinski sign (extensor plantar response)
- No muscle wasting (unlike LMN)
6. Anterior Horn Cells (not a tract, but critical)
These are the lower motor neurons (LMN) sitting in the anterior grey matter.
Damage → Lower Motor Neuron syndrome:
- Ipsilateral segmental flaccid weakness
- Decreased/absent reflexes
- Muscle wasting and fasciculations
- No Babinski
- Seen in: polio, motor neuron disease (at level of lesion in ALS)
SUMMARY TABLE - TRACTS AT A GLANCE
| Tract | Location | Carries | Crosses where | Side of deficit |
|---|---|---|---|---|
| Posterior column | Dorsal | Vibration, proprioception, fine touch | Medulla | Ipsilateral |
| Lateral spinothalamic | Lateral | Pain, temperature | Anterior commissure (spinal cord) | Contralateral |
| Ventral spinothalamic | Anterior | Crude touch | Spinal cord | Contralateral |
| Spinocerebellar | Lateral | Unconscious proprioception | Mostly ipsilateral | Ipsilateral |
| Lateral corticospinal | Lateral | Voluntary movement | Medulla (already crossed) | Ipsilateral |
| Anterior horn cell | Anterior grey | LMN to muscle | None | Ipsilateral, segmental |
CLINICAL SYNDROMES FROM COMBINED TRACT DAMAGE
Brown-Séquard Syndrome (Hemicord - one side cut)
| Side of lesion | Finding |
|---|---|
| Ipsilateral | UMN weakness (corticospinal) + loss of vibration/proprioception (dorsal column) |
| Contralateral | Loss of pain and temperature 1-2 levels below (spinothalamic crossed) |
Cause: Stab wound, tumour on one side, MS plaque
Central Cord Syndrome (Central canal damaged)
- Pain and temperature fibers as they cross are destroyed
- Arms worse than legs (cervical crossing fibers affected first)
- "Cape/shawl" dissociated sensory loss - pain/temperature lost, touch/vibration preserved
- Causes: Syringomyelia, hyperextension injury in elderly, intrinsic tumour
Anterior Cord Syndrome (Anterior spinal artery occlusion)
| Lost | Preserved |
|---|---|
| Motor (bilateral UMN weakness) | Vibration and proprioception |
| Pain and temperature (bilateral) | (posterior columns spared) |
| Bladder/bowel dysfunction |
Cause: Anterior spinal artery infarct (aortic surgery, aortic dissection)
Posterior Cord Syndrome
| Lost | Preserved |
|---|---|
| Vibration, proprioception (bilateral) | Motor power |
| Sensory ataxia, Romberg positive | Pain and temperature |
Causes: B12 deficiency (subacute combined degeneration), tabes dorsalis, multiple sclerosis
Complete Cord Transection
Everything lost below the level:
- Bilateral UMN weakness → spastic paraplegia/quadriplegia
- All sensation lost below level
- Bladder, bowel, sexual dysfunction
- Initially: spinal shock (flaccid, areflexic) → after days/weeks: spasticity and hyperreflexia return
Conus Medullaris Syndrome (sacral cord)
- Saddle anaesthesia (S3-S5 - perianal, perineal area)
- Bladder/bowel dysfunction (urinary retention + incontinence, lax anal tone)
- Impotence
- Bulbocavernosus and anal reflexes absent
- Leg strength largely preserved (distinguishes from cauda equina)
Cauda Equina Syndrome (nerve roots, not cord)
- Low back + radicular pain (prominent)
- Asymmetric leg weakness and sensory loss
- Variable areflexia
- Relative sparing of bladder/bowel (vs. conus)
- LMN signs (flaccid, wasted)
Quick Memory Aid
Posterior columns = "Same side, Fine sensation, Falls with eyes closed"
Spinothalamic = "Opposite side, Pain & Temp, Crosses in cord"
Corticospinal = "Same side, Motor, UMN signs"
BACK of cord = feel where you are
SIDE of cord = pain/temp (opposite) + movement (same)
FRONT of cord = motor neurons (LMN)
CENTRE = crossing pain fibers → cape distribution
Sources: Harrison's Principles of Internal Medicine 22e (p. 3603-3605); Localization in Clinical Neurology 8e (p. 47-48); Adams & Victor's Principles of Neurology 12e; Ganong's Review of Medical Physiology
Why umn and lmn damage cause ipsilateral loss and why lmn not cause babinski sign
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
I have all the content needed. Here is the complete, clear answer.
Why UMN and LMN Damage Both Cause Ipsilateral Weakness - and Why Only UMN Causes Babinski
Part 1: Why Does Both UMN and LMN Damage Cause Ipsilateral Weakness in the Spinal Cord?
This is a really important question and the key is understanding where the corticospinal tract crosses.
The Motor Pathway - Two Neurons
The voluntary motor system has exactly two neurons:
BRAIN (Motor cortex)
↓
Upper Motor Neuron (UMN)
= corticospinal tract
[crosses at the MEDULLA - pyramidal decussation]
↓
SPINAL CORD (Anterior horn)
↓
Lower Motor Neuron (LMN)
= alpha motor neuron
[exits via ventral root - NEVER crosses]
↓
MUSCLE
The Key Point: The Tract Has ALREADY Crossed Before It Enters the Cord
The corticospinal tract (the UMN axon) decussates (crosses) at the pyramidal decussation in the medulla - BEFORE it descends into the spinal cord.
So by the time it is travelling down the spinal cord, it is already on the opposite side from the brain's motor cortex - it is now the same side (ipsilateral) as the limb it controls.
This means:
| Location of damage | Side of weakness |
|---|---|
| Above the decussation (motor cortex, internal capsule, cerebral peduncle, upper medulla) | Contralateral (opposite side to lesion) |
| Below the decussation (spinal cord - UMN damage in cord) | Ipsilateral (same side as lesion) |
| LMN (anterior horn, ventral root, nerve) | Ipsilateral (same side, same segment) |
"Motor cortex or medulla (above the pyramidal decussation): contralateral to weakness. Cervical spinal cord (below the pyramidal decussation): ipsilateral to weakness."
- Neuroanatomy through Clinical Cases 3rd Ed.
Diagram
LEFT Motor Cortex
|
| [UMN descends in LEFT internal capsule]
|
MEDULLA ←—— PYRAMIDAL DECUSSATION ——→
| (fibres CROSS here)
|
RIGHT lateral corticospinal tract
| [now on the RIGHT side of the cord]
|
RIGHT anterior horn cell (LMN)
|
RIGHT limb muscles
So a stroke in the LEFT motor cortex → RIGHT hemiplegia (contralateral - above decussation)
But a spinal cord lesion on the RIGHT → RIGHT limb weakness (ipsilateral - below decussation, already crossed)
And an LMN lesion (e.g., right L4 root) → RIGHT leg weakness (ipsilateral - LMN never crosses at all)
Why Does LMN Damage Also Cause Ipsilateral Weakness?
Because the LMN (anterior horn cell and its axon) never crosses at all. It goes directly from the ventral horn of the spinal cord straight out to the muscle on the same side. There is no crossing in the LMN. So damaging it simply knocks out the muscles it supplies on the same side.
Part 2: Why Does UMN Damage Cause Babinski Sign, But LMN Damage Does NOT?
This is the most elegant question, and the answer requires understanding what the Babinski sign actually is.
What Is the Babinski Sign?
When you stroke the lateral sole of the foot:
- Normal adult response (LMN intact, UMN intact): All toes flex downward (plantar flexion) - negative Babinski
- UMN lesion response: The big toe dorsiflexes (extends upward) and the other toes fan out - positive Babinski
"The Babinski sign is dorsiflexion of the great toe and fanning of the other toes when the lateral aspect of the sole of the foot is scratched. In adults, the normal response to this stimulation is plantar flexion in all the toes."
- Ganong's Review of Medical Physiology
What the Babinski Sign Actually Represents
The Babinski sign is NOT a new reflex. It is actually a primitive nociceptive (withdrawal) reflex that is:
- Present normally in infants under 1 year (corticospinal tract is not yet myelinated)
- Suppressed in normal adults by the descending inhibitory influence of the corticospinal tract
It is a "disinhibited flexion withdrawal reflex" - the foot and toes are trying to withdraw from a painful stimulus by dorsiflexing (which in the lower limb = extension of the big toe upward).
"The Babinski sign... a disinhibited flexion withdrawal reflex, characterized by dorsiflexion (extension) of the large toe... Such response is considered normal until the age of 1 year."
- Localization in Clinical Neurology 8e
Why UMN Damage Releases the Babinski
In a normal adult, the corticospinal tract tonically inhibits this primitive withdrawal reflex. It keeps the spinal reflex circuits under cortical control.
When the UMN (corticospinal tract) is damaged:
- This inhibitory control is removed
- The primitive spinal reflex arc is released (disinhibited)
- The spinal cord reverts to its primitive withdrawal pattern
- The result: big toe extends upward → positive Babinski
Normal adult:
Corticospinal tract ——INHIBITS——→ Primitive withdrawal reflex
(kept suppressed)
Result: Normal plantar flexion (toes curl down)
UMN lesion:
Corticospinal tract DAMAGED → inhibition REMOVED
↓
Primitive reflex RELEASED
Result: Babinski sign (big toe up, toes fan)
Why LMN Damage Does NOT Cause Babinski
For a Babinski sign to appear, the spinal reflex arc must be intact - you need:
- Intact sensory nerve (afferent signal from sole of foot)
- Intact spinal cord interneurons
- Intact LMN (alpha motor neuron) + intact neuromuscular junction + intact muscle
- UMN must be absent/damaged (to remove inhibition)
When the LMN is damaged, the final common pathway is broken. Even though the inhibition from the UMN is gone (so the primitive reflex COULD be disinhibited), the motor neuron or its axon cannot transmit the signal to the muscle. The reflex arc is physically interrupted.
It is like having the brakes removed on a car (UMN gone) but the engine is also dead (LMN gone) - the car still doesn't move.
LMN lesion:
Sole stimulus → sensory nerve → spinal cord interneuron
↓
Alpha motor neuron DAMAGED
↓
No signal reaches muscle
↓
NO REFLEX AT ALL (areflexia)
NO Babinski possible
Complete Comparison Table: UMN vs LMN
| Feature | UMN Lesion | LMN Lesion |
|---|---|---|
| Location | Motor cortex, internal capsule, brainstem (above decussation) OR corticospinal tract in spinal cord (below decussation) | Anterior horn cell, ventral root, peripheral nerve, NMJ |
| Tone | Increased (spasticity) | Decreased (flaccidity) |
| Weakness | Present | Present |
| Reflexes | Hyperreflexia (exaggerated) | Areflexia (absent) |
| Babinski sign | Positive (big toe up) | Absent (reflex arc broken) |
| Muscle wasting | Late/minimal | Early and prominent |
| Fasciculations | Absent | Present (dying LMN fires randomly) |
| Clonus | Present (in severe cases) | Absent |
| Side of deficit | Ipsilateral in cord; contralateral if above medullary decussation | Ipsilateral always |
| Example diseases | Stroke, MS, spinal cord compression, MND (UMN component) | Polio, Guillain-Barré, disc herniation, MND (LMN component) |
Why Reflexes Are Hyperactive in UMN But Absent in LMN
Stretch reflex (deep tendon reflex) circuit:
Tendon tap → muscle stretch → muscle spindle (Ia fibre)
→ enters dorsal horn → DIRECTLY synapses on alpha motor neuron (LMN)
→ LMN fires → muscle contracts (reflex)
This is a monosynaptic arc - it lives entirely in the spinal cord and peripheral nerve.
-
UMN damage: The arc is intact. But the descending inhibitory signals that normally dampen the sensitivity of this arc are gone. The arc becomes overactive → hyperreflexia, clonus. The muscle spindle is now set at higher sensitivity.
-
LMN damage: The alpha motor neuron (the LMN) IS the reflex arc. Damage to it means the arc is physically broken - no matter how strongly the spindle fires, there is no motor neuron to respond → areflexia.
Sources: Ganong's Review of Medical Physiology 26e (Clinical Box 12-5: Lower vs Upper Motor Neuron Damage); Localization in Clinical Neurology 8e (p. 34); Neuroanatomy through Clinical Cases 3e (p. 785-787)
Relation between lateral horn of spinal tract and Horner syndrome
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I have everything - rich textbook content and two excellent images. Here is the complete answer.
The Lateral Horn, the Sympathetic Pathway, and Horner Syndrome
The Lateral Horn - What Is It?
The spinal cord grey matter has three horns:
| Horn | Location | Contains |
|---|---|---|
| Dorsal (posterior) horn | Back | Sensory interneurons |
| Ventral (anterior) horn | Front | Lower motor neurons (alpha motor neurons) |
| Lateral horn | Side | Preganglionic autonomic neurons |
The lateral horn (also called the intermediolateral cell column) is present only at spinal levels T1 to L2/L3 - these are the thoracolumbar segments. It contains the preganglionic sympathetic neurons - the cell bodies of the first neuron in the entire sympathetic nervous system.
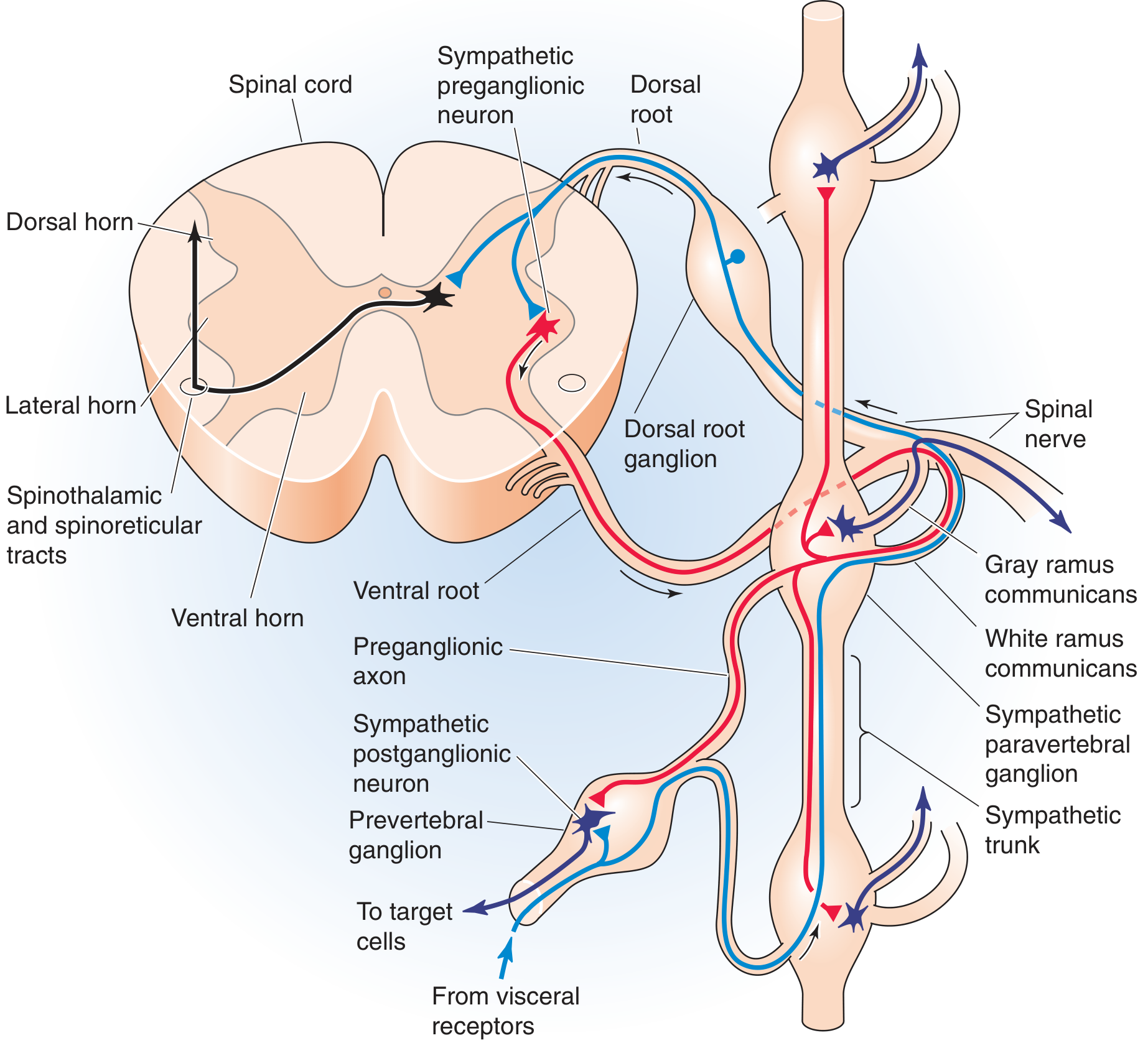
The lateral horn preganglionic neuron (red) exits via the ventral root → white ramus communicans → paravertebral ganglion. Postganglionic neuron (blue) travels to target organs.
"The cell bodies of preganglionic sympathetic motor neurons are located in the thoracic and upper lumbar spinal cord between levels T1 and L3. At these spinal levels, autonomic neurons lie in the intermediolateral cell column, or lateral horn, between the dorsal and ventral horns."
- Medical Physiology (Boron & Boulpaep)
The Three-Neuron Sympathetic Pathway to the Eye
The sympathetic supply to the eye and face travels through a 3-neuron chain. All three neurons must be intact for normal sympathetic function. Damage to any one of them causes Horner syndrome.
Neuron 1 - Central Neuron (First-Order)
- Origin: Hypothalamus (posterior hypothalamus)
- Path: Descends through the brainstem (tegmentum) → lateral reticulospinal tract of the spinal cord
- Destination: Synapse in the lateral horn at C8-T2 (the ciliospinal centre of Budge)
- Never crosses - stays ipsilateral throughout
Neuron 2 - Preganglionic Neuron (Second-Order) - THE LATERAL HORN NEURON
- Origin: Lateral horn at C8-T1-T2 (ciliospinal centre)
- Path: Exits via ventral root → enters white ramus communicans → travels over the apex of the lung → over the subclavian artery → ascends the neck alongside the carotid artery
- Destination: Synapses in the superior cervical ganglion (in the neck, at level of C2-C3)
- This is where the T1 lateral horn neuron is most relevant to Horner syndrome
Neuron 3 - Postganglionic Neuron (Third-Order)
- Origin: Superior cervical ganglion
- Path: Travels along the internal carotid artery → into the cavernous sinus → joins the ophthalmic branch of the trigeminal nerve (V1) → enters the orbit
- Destination: Innervates:
- Pupillary dilator muscle (iris)
- Müller's muscle (upper eyelid elevator - smooth muscle)
- Lower lid retractor (inferior tarsal muscle)
- Sweat glands of the face (via external carotid → facial branches)
The Connection: How Lateral Horn Damage Causes Horner Syndrome
The T1 lateral horn (second-order neuron) is the key spinal cord link. When it or any part of this 3-neuron chain is interrupted, the eye and face lose their sympathetic supply.
The normal sympathetic actions on the eye are:
- Dilates the pupil (radial/dilator muscle of iris)
- Elevates the upper eyelid (Müller's smooth muscle)
- Elevates the lower lid (inferior tarsal muscle)
- Stimulates sweating on same side of face
- Causes slight protrusion of the eyeball
When this chain is cut - at ANY level - you lose all these actions. The parasympathetic tone is now unopposed, and the result is the classic triad:
Horner Syndrome - The Classic Triad
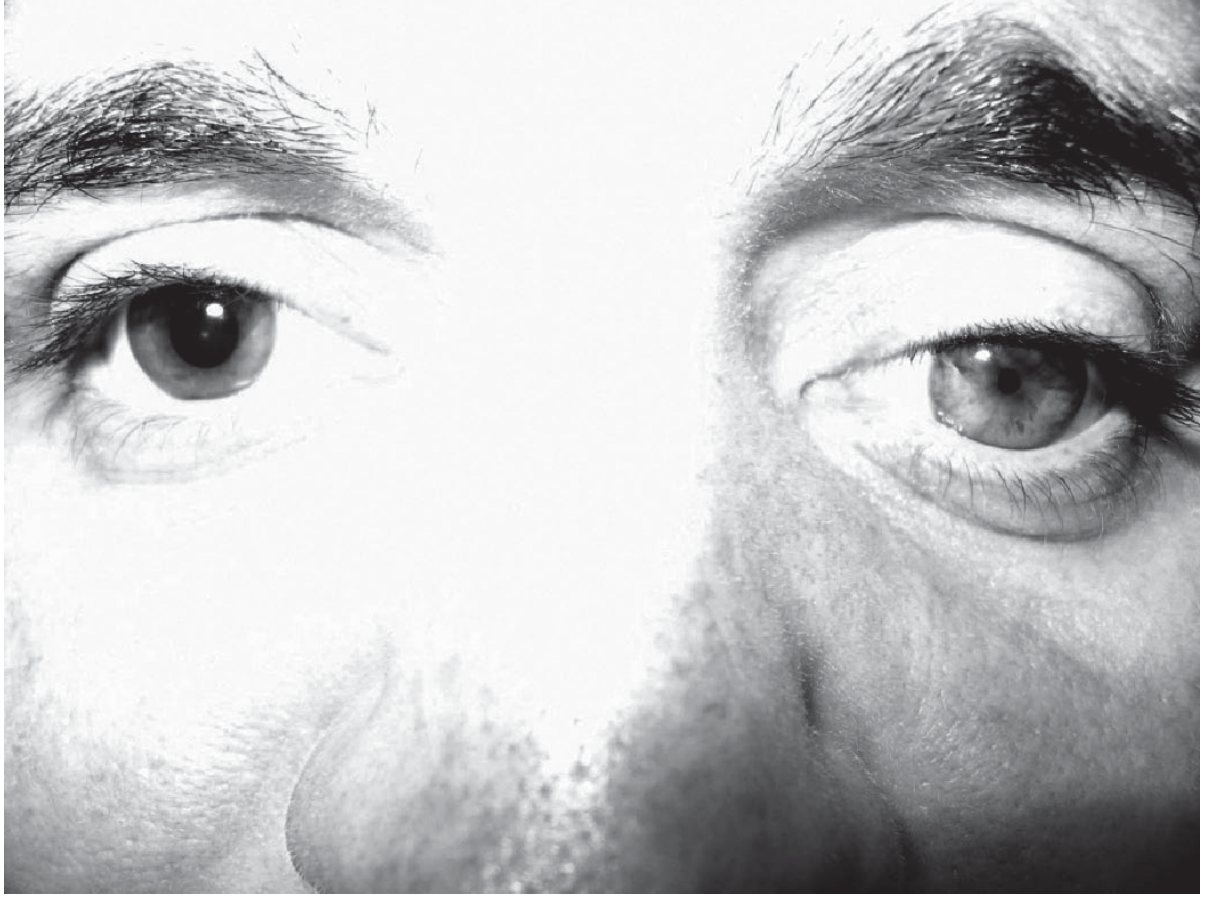
Congenital Horner syndrome (left side): miosis + ptosis + heterochromia iridis
| Sign | Mechanism | Structure lost |
|---|---|---|
| Miosis (small pupil) | Dilator pupillae muscle paralysed; constrictor (parasympathetic) unopposed | Sympathetic → dilator |
| Ptosis (drooping upper lid) | Müller's smooth muscle (upper lid) paralysed | Sympathetic → Müller's muscle |
| Anhidrosis (no sweating) | Loss of sympathetic sudomotor supply to ipsilateral face | Sympathetic → sweat glands |
| (Enophthalmos) | Apparent sunken eye - actually an illusion from narrowed palpebral fissure | - |
| (Lower lid elevation) | "Reverse ptosis" - inferior tarsal muscle paralysed, lower lid rises slightly | Sympathetic → inferior tarsal |
Important: The anisocoria (unequal pupils) is more obvious in the dark (unlike a 3rd nerve palsy which is worse in light). This is because in the dark the normal pupil dilates widely but the Horner pupil cannot dilate - the difference becomes exaggerated.
Where Along the 3-Neuron Chain Can Damage Occur?
HYPOTHALAMUS
↓ (Neuron 1 - Central)
BRAINSTEM → LATERAL RETICULOSPINAL TRACT
↓
LATERAL HORN at C8-T1-T2
↓ (Neuron 2 - Preganglionic - exits lateral horn)
Over apex of lung → over subclavian artery
↓
SUPERIOR CERVICAL GANGLION (neck)
↓ (Neuron 3 - Postganglionic)
Along internal carotid → cavernous sinus → orbit
↓
PUPIL DILATOR + MÜLLER'S MUSCLE + SWEAT GLANDS
| Level of Damage | Neuron | Common Causes |
|---|---|---|
| Hypothalamus or brainstem | 1st order (central) | Stroke, MS, lateral medullary infarct (Wallenberg syndrome), tumour |
| Spinal cord (C8-T2 lateral horn) | 2nd order (preganglionic) | Syringomyelia, spinal cord trauma, MS, Brown-Séquard |
| Apex of lung / T1 root | 2nd order | Pancoast tumour (apical lung cancer - most classic cause), cervical rib |
| Neck (sympathetic chain) | 2nd order | Neck surgery, carotid dissection, lymph nodes, trauma |
| Along internal carotid | 3rd order | Carotid artery dissection, cavernous sinus thrombosis |
| Cavernous sinus | 3rd order | Cavernous sinus lesion, aneurysm |
Why Spinal Cord Lesions Cause Horner Syndrome
Any lesion affecting the lateral horn at C8-T1-T2 or the descending first-order sympathetic fibres in the lateral cord causes ipsilateral Horner syndrome:
- Syringomyelia: the expanding central cavity erodes outward to the lateral horn
- Spinal cord trauma at cervicothoracic level
- Brown-Séquard syndrome: the hemicord lesion also damages lateral horn + descending sympathetics
- Multiple sclerosis plaques in the lateral cord
- Hemisection of the cord at any level above T2 disrupts the descending first-order sympathetic pathway
The Horner syndrome from a spinal cord lesion is always ipsilateral to the lesion because the sympathetic pathway never crosses in the spinal cord (it travels ipsilaterally all the way down from the hypothalamus to the T1 lateral horn).
Anhidrosis Pattern Helps Localize the Lesion
| Level of lesion | Pattern of anhidrosis |
|---|---|
| Central (1st order - hypothalamus/brainstem/spinal cord) | Entire ipsilateral face, arm, and trunk |
| Pre-ganglionic (2nd order - T1 to superior cervical ganglion) | Entire ipsilateral face |
| Post-ganglionic (3rd order - along carotid) | No anhidrosis (or only medial forehead/side of nose) - because sudomotor fibres diverge before the superior cervical ganglion |
This anhidrosis pattern is clinically very useful: if a patient has Horner syndrome with NO anhidrosis, the lesion is postganglionic (e.g., carotid dissection).
Quick Summary
Lateral horn (T1-T2) = home of the 2nd-order preganglionic sympathetic neuron
Damage to lateral horn (or anywhere along 3-neuron chain)
↓
Loss of sympathetic supply to ipsilateral eye and face
↓
HORNER SYNDROME:
• Miosis (small pupil) - dilator paralysed
• Ptosis (drooping lid) - Müller's muscle paralysed
• Anhidrosis (no sweat) - sweat glands denervated
Sources: Adams & Victor's Principles of Neurology 12e (p. Horner Syndrome); Neuroanatomy through Clinical Cases 3e (p. 608-609); Medical Physiology - Boron & Boulpaep (p. 502); Ganong's Review of Medical Physiology 26e; Lippincott Illustrated Reviews Pharmacology
Werding Hoffman disease
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
I have enough comprehensive content from Adams & Victor and Robbins. I have the full SMA classification table, molecular genetics, clinical features, differential diagnosis, and treatment. Let me compile the complete answer.
Werdnig-Hoffmann Disease (SMA Type I)
Definition and Classification
Werdnig-Hoffmann disease is Spinal Muscular Atrophy Type I (SMA I) - the most severe form of a group of genetically linked childhood disorders characterised by progressive degeneration of lower motor neurons (anterior horn cells of the spinal cord + motor nuclei of the brainstem), leading to weakness and muscle wasting.
It is:
- The most common lethal autosomal recessive disease of childhood after cystic fibrosis
- A pure LMN disease - the corticospinal tract and upper motor neurons are completely spared
- Named after Guido Werdnig and Johann Hoffmann who described it in the 1890s
Molecular Genetics - The SMN Gene
The key to understanding all SMA types is the SMN (Survival Motor Neuron) gene on chromosome 5q13.
Two copies of the gene exist side by side:
| Gene | Function | Result |
|---|---|---|
| SMN1 | Produces full-length, stable, functional SMN protein | The "working" gene |
| SMN2 | Similar sequence but a key base-pair difference alters mRNA splicing → produces mostly truncated, unstable SMN protein (rapidly degraded) | Backup gene - only ~10-15% functional protein |
What goes wrong in SMA:
"All forms of 5q-SMA are caused by loss-of-function mutations in SMN1 (usually deletions), with the differences in clinical phenotype being determined by the number of SMN2 gene copies."
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- SMN1 is deleted or mutated on both chromosomes (homozygous loss)
- Motor neurons are highly dependent on SMN protein for survival (it is involved in assembly of the spliceosome - the RNA processing machinery)
- Without adequate SMN protein, anterior horn cells progressively die
- The more copies of SMN2 a patient has, the more backup functional protein is made → milder disease
Why Werdnig-Hoffmann (SMA I) is the Worst:
SMA I patients have only 2 copies of SMN2 - the minimum - so they produce the least backup protein and have the most severe anterior horn cell loss.
SMA Classification - All Types at a Glance
| Type | Name | SMN2 copies | Age of onset | Clinical features | Prognosis |
|---|---|---|---|---|---|
| SMA I | Werdnig-Hoffmann | 2 | Preterm to 6 months | Floppy baby, unable to sit, weak suck/swallow, may have arthrogryposis | Few survive 1 year |
| SMA II | Dubowitz disease | ≥3 | 6-15 months | Proximal weakness, fasciculation, fine hand tremor, unable to stand | Death from respiratory complications |
| SMA III | Kugelberg-Welander | ≥3 | 1 yr to adolescence | Delayed motor development, proximal leg weakness | Slowly progressive, variable |
| SMA IV | Adult onset | ≥4 | After age 30 | Proximal limb + diaphragm weakness | Slowly progressive; wheelchair eventually |
| Kennedy syndrome | Bulbospinal atrophy | - | Early adulthood | Scapuloperoneal/distal atrophy, bulbar weakness, gynecomastia | Slowly progressive |
Pathology
- Selective degeneration of anterior horn cells of the entire spinal cord (especially cervical and lumbar enlargements)
- Motor nuclei of the brainstem (especially CN IX, X, XII) also affected
- Spinal cord looks shrunken; ventral roots are thin
- Denervation atrophy of skeletal muscles - groups of atrophic fibres next to normal "re-innervated" fibres (grouped atrophy)
- The corticospinal tracts, dorsal columns, and sensory neurons are entirely spared - this is a pure LMN disease
- Arthrogryposis (fixed joint contractures) may occur in utero because of absent fetal movement
Clinical Features of SMA I (Werdnig-Hoffmann)
Onset: Birth to 6 months (most before 3 months; some noted in utero as reduced fetal movements)
The "Floppy Baby"
The hallmark presentation is the floppy infant (hypotonia):
- Baby lies in a characteristic "frog-leg" posture - hips abducted, knees flexed, arms limp at sides
- Profound generalised flaccid weakness - limbs feel like "wet rags"
- Head lag - head drops back completely on pull-to-sit
LMN Signs (all are LMN - NO UMN features)
| Sign | Finding |
|---|---|
| Tone | Severely reduced (hypotonia, flaccidity) |
| Reflexes | Absent (areflexia) - deep tendon reflexes cannot be elicited |
| Babinski | Absent - no Babinski sign (LMN arc broken) |
| Fasciculations | Present - visible in tongue (characteristic), sometimes limb muscles |
| Muscle wasting | Severe, progressive |
| Weakness | Proximal > distal; legs > arms |
Bulbar Involvement (brainstem motor nuclei)
- Weak suck - poor feeding, difficulty latching
- Weak swallow - aspiration risk
- Tongue fasciculations - highly characteristic finding, nearly diagnostic
- Weak cry - feeble, high-pitched cry
- Facial expression relatively preserved (facial muscles less involved)
Respiratory
- Intercostal muscle weakness → paradoxical breathing (belly breathes in while chest sinks in - "chest caving on inspiration")
- Diaphragm relatively spared early → characteristic bell-shaped chest (narrow rib cage, protruding abdomen)
- Progressive respiratory failure is the cause of death
- Recurrent aspiration pneumonia
Sensory and Cognitive - NORMAL
- Sensation is completely intact (sensory neurons not affected)
- Intelligence is normal - these children are bright and alert, which makes the condition heartbreaking
- Eye movements are preserved
What the Child CANNOT Do
- Cannot sit unsupported (defining feature of SMA I)
- Cannot roll
- Cannot hold head up
- Cannot bear weight on legs
Diagnosis
| Investigation | Finding |
|---|---|
| Genetic testing (SMN1 deletion) | Gold standard - homozygous deletion of SMN1 exon 7 confirms diagnosis |
| EMG | Fibrillations, positive sharp waves, fasciculation potentials (denervation); reduced interference pattern |
| Nerve conduction studies | Motor conduction velocities normal or near-normal (axons intact; it is cell bodies dying) |
| Muscle biopsy | Grouped atrophy - large groups of atrophic fibres (denervated) adjacent to hypertrophied fibres (reinnervated) |
| CK (creatine kinase) | Normal or mildly elevated (unlike muscular dystrophy where CK is markedly elevated) |
| Antenatal diagnosis | Possible via amniocentesis/CVS if family history known |
Differential Diagnosis of the "Floppy Baby"
| Condition | Key distinguishing feature |
|---|---|
| SMA I | Tongue fasciculations, areflexia, normal CK, SMN1 deletion |
| Congenital myotonic dystrophy | Mother has myotonia on exam, face diplegia prominent |
| Congenital myopathy (nemaline, central core) | Muscle biopsy shows structural abnormalities |
| Duchenne muscular dystrophy | Markedly elevated CK; presents later (3-5 yrs) |
| Prader-Willi syndrome | Hypotonia + hypogonadism + feeding difficulties; genetic (chromosome 15) |
| Hypothyroidism | Treatable; thyroid function tests diagnostic |
| Birth asphyxia | History, UMN signs may emerge later |
| Hypotonic cerebral palsy | UMN signs emerge; developmental regression with time |
| Guillain-Barré | Ascending weakness, CSF protein elevated |
| Botulism (infant) | Descending paralysis, dilated fixed pupils, exposure to honey |
Treatment
Previously: No effective treatment - supportive only
- Nutritional support (nasogastric/PEG feeding)
- Physiotherapy, splinting to prevent contractures
- Respiratory support (BiPAP, eventually ventilation)
- Palliative care discussion
Now: Three Disease-Modifying Treatments (Revolutionary)
"Two novel treatments have been remarkable developments in this otherwise dismal field."
- Adams & Victor's Principles of Neurology
| Treatment | Mechanism | Route |
|---|---|---|
| Nusinersen (Spinraza) | Antisense oligonucleotide - modifies SMN2 mRNA splicing to produce more full-length functional SMN protein | Intrathecal injection (repeated) |
| Onasemnogene abeparvovec (Zolgensma) | Gene therapy - AAV9 vector delivers a functional copy of SMN1 directly to motor neurons | Single intravenous infusion (~$2 million per dose) |
| Risdiplam (Evrysdi) | Small molecule - modifies SMN2 splicing (similar to nusinersen but oral) | Oral daily |
Key: These treatments are most effective when given before symptom onset (pre-symptomatic), which is why newborn screening for SMA is now recommended.
Prognosis of SMA I (Without Treatment)
- Most infants die within the first year of life
- Death is from respiratory failure - typically due to recurrent pneumonia and inability to clear secretions
- Virtually none survive beyond 2 years without ventilatory support
- With maximal supportive care (ventilation), survival can be extended
With modern gene therapy started early: Dramatically improved outcomes; some children treated pre-symptomatically are achieving near-normal motor milestones.
Why It Is a Pure LMN Disease - Summary
SMN1 gene deleted (chromosome 5q13)
↓
Insufficient SMN protein
↓
Anterior horn cells (and brainstem motor nuclei) degenerate
↓
No signal reaches muscles (final common pathway dead)
↓
Flaccid paralysis + areflexia + fasciculations + wasting
(ALL LMN signs - NO UMN signs whatsoever)
↓
Respiratory muscles fail → respiratory failure → death
Sources: Adams & Victor's Principles of Neurology 12e (Table 38-7, SMA classification + Werdnig-Hoffmann clinical section); Robbins, Cotran & Kumar Pathologic Basis of Disease (SMA/SMN genetics); Campbell's Operative Orthopaedics 15e
This is a shared conversation. Sign in to Orris to start your own chat.