Parkinsons
Reading File
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND "treatment"[MeSH Terms]
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full response.
Parkinson's Disease
Overview
Parkinson's disease (PD) is the second most common neurodegenerative disorder after Alzheimer's disease, affecting approximately 1 in 1000 in the general population and at least 1% of persons over 65. Men are affected slightly more often than women (3:2 ratio). First described by James Parkinson in 1817, it is a progressive disorder primarily of dopaminergic neurons in the substantia nigra pars compacta. - Goldman-Cecil Medicine, p. 3961
Pathobiology
Etiology
PD results from a combination of genetic and environmental factors:
-
Environmental risks: Pesticides, heavy metals, traumatic brain injury; the neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) directly models the disease by inhibiting complex I of the mitochondrial electron transport chain, causing selective dopaminergic neuron death via oxidative stress.
-
Genetic causes (~10% of cases):
- Autosomal dominant: SNCA (α-synuclein gene - point mutations, duplications/triplications), LRRK2 (the most common cause of familial PD), VPS35
- Autosomal recessive (early-onset): Parkin (PARK2), PINK1, DJ-1
- Risk factor: Glucocerebrosidase (GBA) heterozygous mutations (Gaucher disease gene) - associated with faster progression and higher dementia risk
-
Prion-like spread: Cell-to-cell transmission of misfolded α-synuclein via "permissive templating" propagates neurodegeneration through the nervous system.
-
Goldman-Cecil Medicine, p. 3962; Robbins & Kumar Basic Pathology, p. 854; Adams and Victor's Principles of Neurology
Pathology
Gross: Pallor of the substantia nigra and locus coeruleus (loss of neuromelanin-containing neurons).
Microscopic:
- Loss of pigmented dopaminergic neurons in substantia nigra pars compacta with gliosis
- Lewy bodies - the pathological hallmark: round, eosinophilic cytoplasmic inclusions consisting of fine filaments of α-synuclein, neurofilaments, and ubiquitin
- Lewy neurites - dystrophic neuritic processes also containing aggregated α-synuclein
Approximately 60% of dopaminergic neurons must degenerate before classic motor features emerge. - Goldman-Cecil Medicine, p. 3962
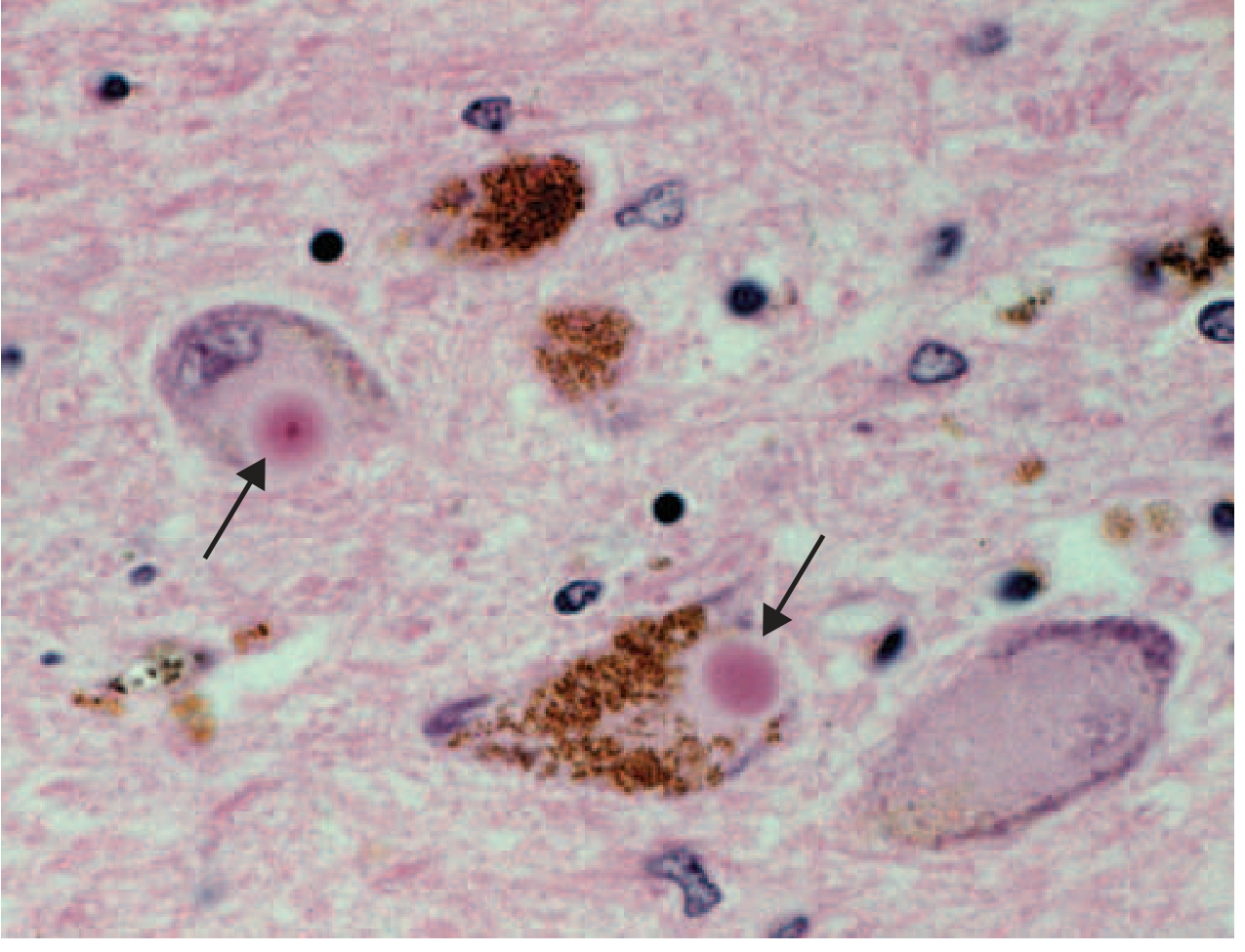
Lewy bodies (arrows) within melanized dopamine neurons in PD (H&E stain). The round eosinophilic inclusions are the pathological hallmark of the disease. - Harrison's Principles of Internal Medicine
Pathologic changes extend beyond the substantia nigra to:
- Brain stem nuclei (dorsal motor nucleus of vagus - early involvement causing sleep/autonomic features)
- Olfactory system (early anosmia)
- Enteric nervous system
- Cortical regions (leading to dementia)
Clinical Features
Prodromal / Premotor Phase
Years to decades before motor symptoms, patients may have:
- REM sleep behavior disorder (RBD) - often earliest manifestation
- Anosmia (loss of smell)
- Autonomic dysfunction (constipation, orthostatic hypotension)
- Depression / anxiety
- Cardiac denervation (reduced MIBG uptake)
These are now recognized as likely representing early disease, not merely "risk factors." - Harrison's Principles of Internal Medicine
Cardinal Motor Features (TRAP)
| Feature | Description |
|---|---|
| Tremor | Resting "pill-rolling" tremor, 4-6 Hz; subsides with movement; accentuated by mental stress; present in only ~20% at onset |
| Rigidity | "Cogwheel" (with tremor) or "lead pipe" (without); increased tone on passive movement; activated by contralateral limb movement |
| Akinesia/Bradykinesia | Slowness, reduced amplitude, fatigue; causes: masked facies (hypomimia), hypophonia, micrographia, reduced arm swing, shuffling gait, freezing of gait, sialorrhea |
| Postural instability | Flexed posture; impaired postural reflexes; festination, propulsion/retropulsion; positive pull test |
Symptoms typically begin unilaterally and this asymmetry often persists. - Goldman-Cecil Medicine, p. 3962; Robbins & Kumar, p. 854
Non-Motor Features
-
Cognitive: Dementia (common in advanced disease); if within 1 year of motor onset → Lewy body dementia
-
Psychiatric: Visual hallucinations (especially drug-related), depression, anxiety, impulse control disorders (with dopamine agonists), paranoid psychosis
-
Autonomic: Orthostatic hypotension, constipation, urinary dysfunction, weight loss
-
Sleep: RBD, excessive daytime sleepiness
-
Pain and sensory symptoms (some levodopa-responsive)
-
Dysphagia: Prevalence 35-82% depending on objective vs. subjective measures; involves oral, pharyngeal, and esophageal phases; aspiration in 15-56%, silent aspiration in 15-33%
-
Goldman-Cecil Medicine (Table 378-2); Bradley and Daroff's Neurology
Diagnosis
Historically based on the UK Brain Bank Criteria (2 of 3 features: tremor, rigidity, bradykinesia) with a 24% postmortem error rate. Revised criteria raised pathological concordance to >90% by emphasizing: bradykinesia + rigidity, asymmetry, and good levodopa response.
The current MDS Clinical Diagnostic Criteria (Movement Disorder Society) require:
- Core feature: Motor parkinsonism (bradykinesia + rigidity ± tremor)
- Supportive criteria (increase confidence): Dramatic response to levodopa, levodopa-induced dyskinesia, rest tremor of a limb, anosmia, cardiac sympathetic denervation
- Absolute exclusion criteria
- Red flags (must be counterbalanced by supportive criteria)
Two levels: Clinically established PD and Clinically probable PD. - Harrison's Principles of Internal Medicine, p. 3539
Dopamine imaging (PET or SPECT - DAT scan): Reduced and asymmetric uptake in posterior putamen with relative caudate sparing - helps distinguish PD from essential tremor and drug-induced parkinsonism.
Differential Diagnosis: Atypical Parkinsonisms ("Parkinson-plus")
| Disorder | Key Distinguishing Features | Levodopa Response |
|---|---|---|
| Multiple System Atrophy (MSA) | Early dysautonomia, cerebellar signs, stridor; MRI: "hot cross bun sign" in pons (MSA-C), striatal changes (MSA-P) | Initial partial response only |
| Progressive Supranuclear Palsy (PSP) | Supranuclear gaze palsy (downward first), axial > limb rigidity, early falls; MRI: "hummingbird sign" | Minimal |
| Corticobasal Degeneration (CBD) | Asymmetric apraxia, cortical sensory loss, alien limb phenomenon, aphasia | Negligible |
| Dementia with Lewy Bodies (DLB) | Fluctuating cognition, recurrent visual hallucinations, RBD; dementia precedes or concurrent with motor symptoms | Variable |
| Vascular parkinsonism | "Lower body" parkinsonism, gait prominent, MRI white matter changes | Poor |
| Drug-induced | Symmetrical, abrupt onset, related to dopamine antagonists (haloperidol, metoclopramide, prochlorperazine) | Resolves on withdrawal |
Other red flags pointing away from PD: early prominent dysarthria/dysphagia, nystagmus, ataxia, bilateral symmetric onset, failure to respond to adequate levodopa trial. - Goldman-Cecil Medicine (Table 378-4); Neuroanatomy through Clinical Cases
Treatment
When to Start
Early treatment - even with mild symptoms - can preserve quality of life. There is no evidence supporting delay of levodopa. - Goldman-Cecil Medicine
Medical Therapy
1. Levodopa + Carbidopa (most effective agent)
- Levodopa is a dopamine precursor that crosses the blood-brain barrier; carbidopa is a peripheral decarboxylase inhibitor that prevents conversion outside the CNS, reducing GI and cardiovascular side effects and increasing CNS availability.
- Motor complications develop in ~50% within 2-5 years:
- Wearing off - end-of-dose deterioration; managed by more frequent dosing, controlled-release formulations, COMT or MAO-B inhibition
- Dyskinesias (peak-dose: chorea/athetosis; diphasic; off-period dystonia) - earlier in young-onset PD
- On-off fluctuations - unpredictable; may benefit from continuous enteral infusion or subcutaneous levodopa prodrugs
2. Dopamine Agonists (pramipexole, ropinirole, rotigotine)
- Preferred as initial therapy in younger (<65), cognitively intact patients to delay motor complications
- Less effective than levodopa; risk of hallucinations, impulse control disorders, excessive daytime sleepiness
- All patients eventually require levodopa addition
3. MAO-B Inhibitors (selegiline, rasagiline, safinamide)
- Prevent dopamine breakdown; useful early (mild symptoms) or as adjunct to reduce wearing-off
- Rasagiline used early when there is little functional impairment
4. COMT Inhibitors (entacapone, opicapone)
- Block peripheral dopamine metabolism; used adjunctively to smooth levodopa action and reduce wearing-off
5. Other agents (less commonly used)
-
Amantadine: Anticholinergic + antiglutamatergic + pro-dopaminergic; useful for dyskinesia
-
Anticholinergics: For tremor-predominant disease in younger patients (cognitive risk limits use)
-
Neuroanatomy through Clinical Cases, p. 792; Goldman-Cecil Medicine, p. 3963; Textbook of Family Medicine
Surgical Treatment: Deep Brain Stimulation (DBS)
Indicated for patients with motor fluctuations/dyskinesias despite optimized medical therapy. Electrodes are placed in either:
- Subthalamic nucleus (STN): Larger motor benefit in "off" state; allows larger medication dose reduction; slightly higher neuropsychiatric risk
- Globus pallidus internus (GPi): Better dyskinesia suppression; more flexible long-term; safer neuropsychiatric profile; preferred for "brittle" dyskinesias
Both targets have shown similar overall motor outcomes in RCTs. DBS does not cure PD but significantly reduces "off" time and dyskinesias. - Bradley and Daroff's Neurology, p. 652
Non-Pharmacological
- Physical therapy: Gait and balance training, fall prevention
- Occupational therapy: Activities of daily living
- Speech therapy: For hypophonia and dysphagia (LSVT LOUD)
- Aerobic exercise: Evidence for positive disease-modifying effect
- Swallowing evaluation: Videofluoroscopy or FEES recommended given high rate of silent aspiration
Prognosis
Disease typically progresses over 10-15 years, eventually causing severe motor slowing approaching near-immobility. Death most commonly results from aspiration pneumonia or trauma from falls. Later-stage features (freezing of gait, postural instability, dementia, dysautonomia) become treatment-resistant over time. - Robbins & Kumar Basic Pathology, p. 854
Sources: Goldman-Cecil Medicine (International Edition); Harrison's Principles of Internal Medicine 22e; Robbins & Kumar Basic Pathology; Neuroanatomy through Clinical Cases (3rd ed.); Bradley and Daroff's Neurology in Clinical Practice; Adams and Victor's Principles of Neurology (12th ed.); Textbook of Family Medicine (9th ed.)
Parkinson pathogenesis
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND "alpha-synuclein"[MeSH Terms] AND pathogenesis
Finding Sources
Reading Article
PMID: 38245249
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Finding Sources
Reading File
Loading Image
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
I now have comprehensive, multi-source textbook data plus the key images and a recent Lancet review. Let me compile the full pathogenesis response.
Parkinson's Disease - Pathogenesis
The pathogenesis of PD is multifactorial, involving interconnected mechanisms that converge on the progressive death of dopaminergic neurons in the substantia nigra pars compacta (SNc). No single dominant cause has been definitively established; rather, these pathways form a pathogenic network in which any single insult may be insufficient alone to cause neurodegeneration - multiple converging hits are likely required. - Harrison's Principles of Internal Medicine 22e
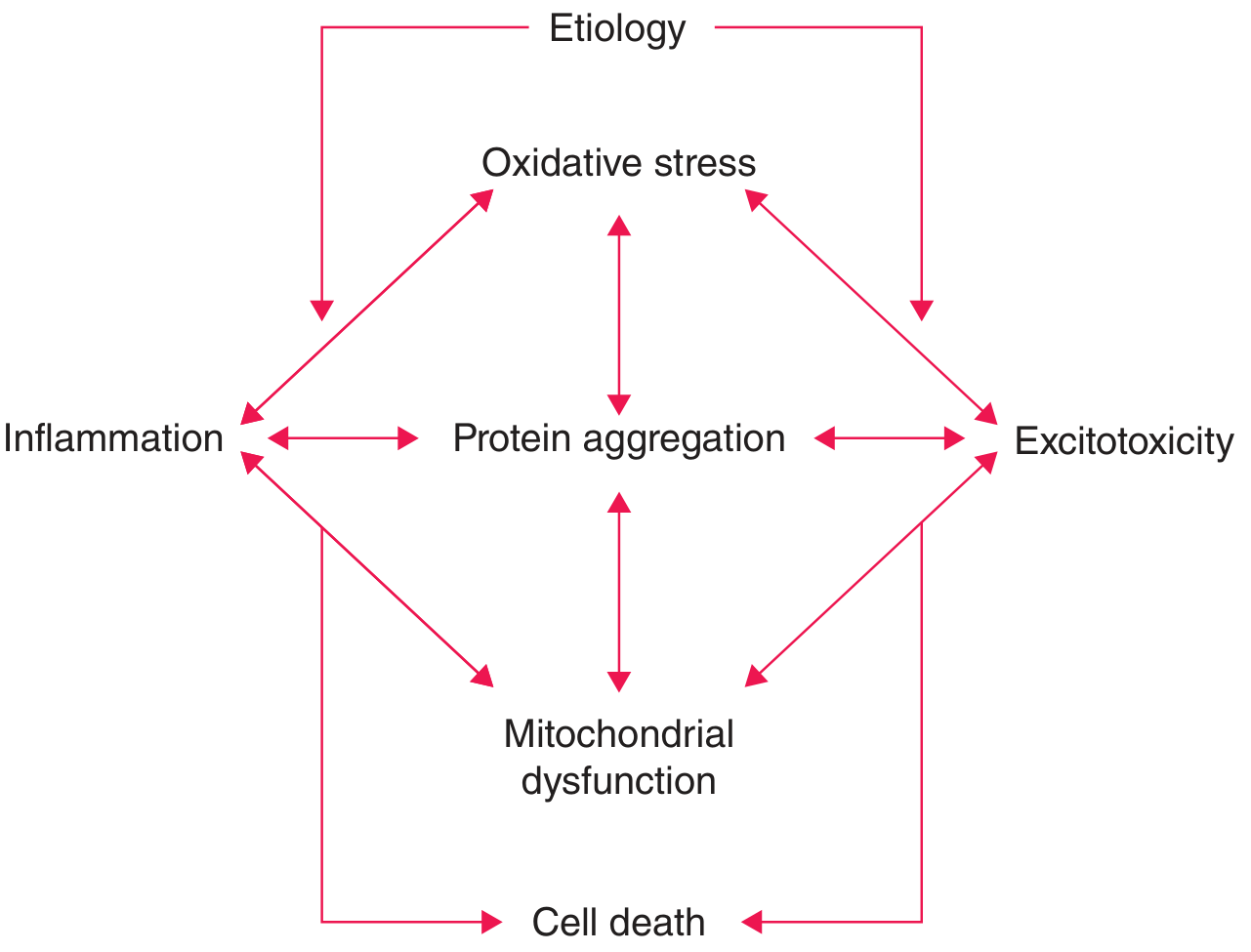
1. α-Synuclein Misfolding and Aggregation
This is the central molecular event in PD pathogenesis.
Normal function: α-Synuclein is a nuclear and synaptic protein involved in synaptic vesicle dynamics. In normal neurons it exists as a soluble, unfolded monomer.
Pathological cascade:
- Under conditions of increased concentration, mutation, or impaired clearance, α-synuclein misfolds into oligomers (protofibrils) - the most cytotoxic form
- Protofibrils polymerize into insoluble fibrils
- Fibrils accumulate in neuronal cytoplasm as Lewy bodies (round eosinophilic inclusions with a dense core and pale halo) and Lewy neurites
- Lewy bodies contain: α-synuclein, ubiquitin, hyperphosphorylated neurofilament proteins, lipids, and iron
Causes of α-synuclein overload or instability:
- SNCA point mutations (A53T, A30P) - promote oligomerization and decrease protein stability
- SNCA duplications and triplications - cause PD by overproducing wild-type protein alone; triplication carriers are more severely affected than duplication carriers
- Impaired proteasomal/lysosomal clearance (see below)
- Interaction with dopamine - α-synuclein forms dopamine adducts that stabilize the toxic protofibrillar form
Why dopaminergic neurons are selectively vulnerable: Their high dopamine turnover generates oxidative metabolites; the protein in high concentration finds conditions that favor aggregation; and their long, unmyelinated axons impose a high metabolic demand.
- Adams and Victor's Principles of Neurology (12th ed.); Robbins, Cotran & Kumar Pathologic Basis of Disease
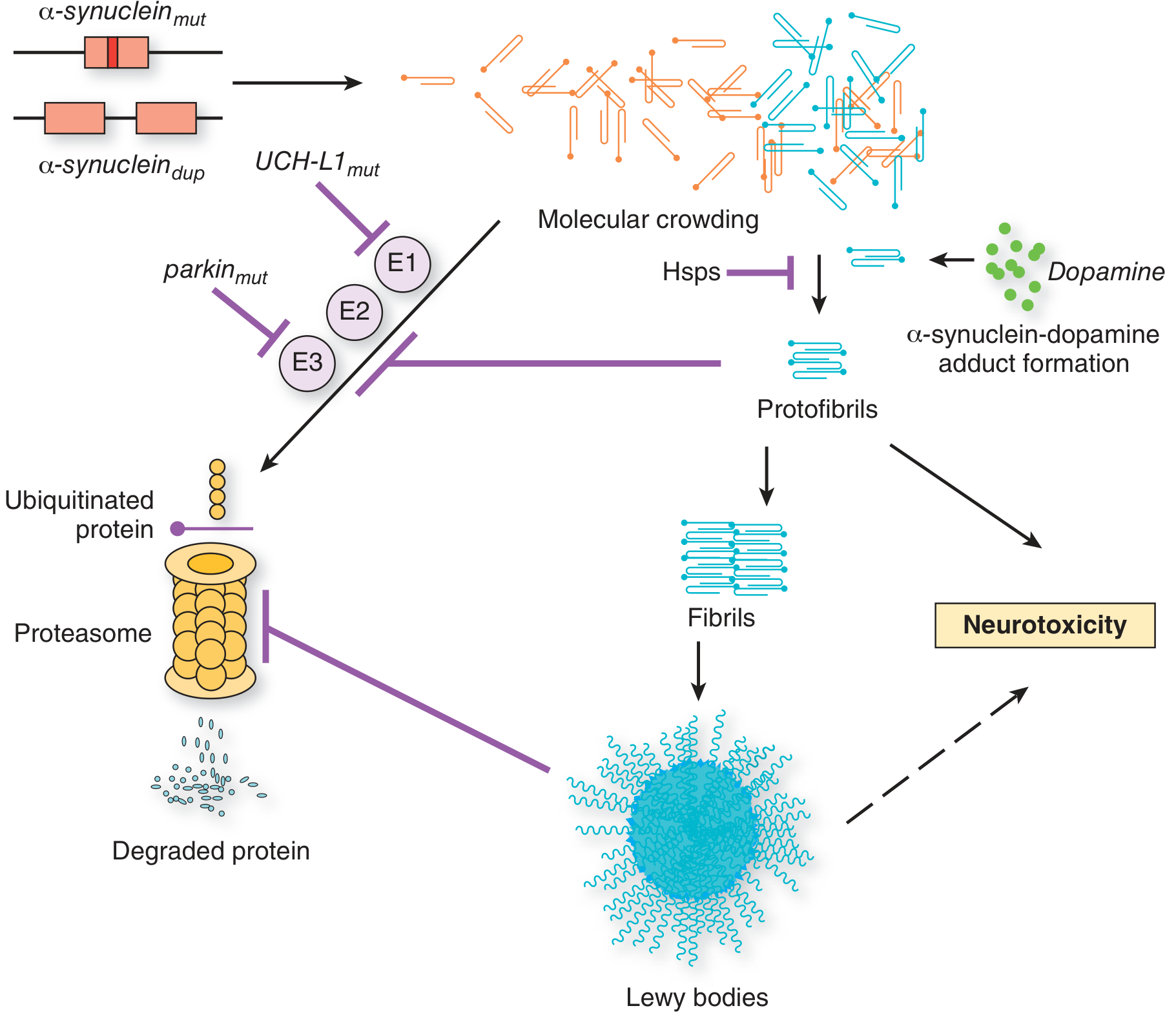
2. Impaired Protein Clearance
Two major degradation systems are involved:
A. Ubiquitin-Proteasome System (UPS)
- Normal pathway: Proteins tagged with ubiquitin (via E1 → E2 → E3 ligase cascade) are degraded by the 26S proteasome
- Parkin (PARK2) is an E3 ubiquitin ligase; parkin mutations impair ubiquitination and proteasomal disposal of α-synuclein and other substrates, causing their accumulation
- UCHL-1 (PARK5): A ubiquitin carboxy-terminal hydrolase; mutations impair the recycling of ubiquitin monomers, compromising the entire UPS
- Importantly, Lewy bodies are not found in most parkin-mutation PD cases, suggesting the Lewy body forms downstream of failed clearance
B. Autophagy-Lysosome Pathway (ALP)
-
α-Synuclein aggregates are primarily cleared via macroautophagy and chaperone-mediated autophagy (CMA)
-
GBA1 (glucocerebrosidase) heterozygous mutations impair lysosomal function → reduced clearance of α-synuclein → accumulation. The reciprocal is also true: accumulated α-synuclein inhibits GCase by interfering with ER-to-Golgi trafficking → vicious cycle
-
GBA1 mutations account for ~5% of PD cases and are associated with faster progression and higher dementia risk
-
This lysosomal impairment is not limited to GBA1-mutation carriers - GCase levels are also reduced in sporadic PD
-
Robbins, Cotran & Kumar, p. 1183-1184; Adams and Victor's, p. 1090; Harrison's Principles of Internal Medicine 22e
3. Mitochondrial Dysfunction
Strong evidence from both genetic and environmental data:
Complex I inhibition:
- MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) selectively destroys dopaminergic neurons via its active metabolite MPP⁺, a potent Complex I inhibitor of the mitochondrial electron transport chain - directly causing oxidative stress and ATP depletion
- Complex I activity is reduced in the SNc and platelets of sporadic PD patients
- Pesticides (rotenone, paraquat) - also Complex I inhibitors - are epidemiologically linked to increased PD risk
Genetic links to mitochondria:
-
PINK1 (PTEN-induced kinase 1): Localizes to the outer membrane of dysfunctional (depolarized) mitochondria; phosphorylates Parkin to recruit it to damaged mitochondria
-
Parkin + PINK1 together execute mitophagy - the autophagic clearance of damaged mitochondria. Mutations in either gene impair mitophagy → accumulation of defective, ROS-generating mitochondria
-
DJ-1 (PARK7): A transcriptional regulator that translocates to mitochondria under oxidative stress and has cytoprotective effects; DJ-1 mutations impair this response and increase α-synuclein aggregation
-
α-Synuclein itself directly interacts with mitochondrial membranes and inhibits Complex I in a dose-dependent manner
-
Bradley and Daroff's Neurology in Clinical Practice; Robbins, Cotran & Kumar, p. 1184
4. Oxidative Stress
Dopaminergic neurons of the SNc are uniquely vulnerable to oxidative stress because:
- Dopamine metabolism (by MAO and auto-oxidation) generates hydrogen peroxide and reactive oxygen species (ROS)
- High iron content of the SNc - iron catalyzes the Fenton reaction, generating hydroxyl radicals
- Reduced glutathione (GSH) levels found early in PD
- Mitochondrial Complex I inhibition further generates superoxide
ROS damages lipids, proteins, and DNA, contributing to neuronal death. Oxidative stress and mitochondrial dysfunction reinforce each other in a bidirectional relationship. - Goldman-Cecil Medicine; Katzung's Basic and Clinical Pharmacology
5. Braak Staging - Spatiotemporal Progression
Braak and colleagues proposed a 6-stage staging system based on α-synuclein immunostaining showing that PD pathology spreads in a predictable caudal-to-rostral pattern:
| Stage | Region Involved | Clinical Correlation |
|---|---|---|
| 1 | Dorsal motor nucleus of vagus; anterior olfactory nucleus | Anosmia, autonomic symptoms (prodromal) |
| 2 | Raphe nuclei; locus coeruleus; reticular formation | REM sleep behavior disorder, depression (prodromal) |
| 3 | Substantia nigra pars compacta (SNc) | Motor symptoms emerge (TRAP) |
| 4 | Mesocortex; thalamus; amygdala | Early cognitive/psychiatric features |
| 5-6 | Neocortex | Dementia, psychosis |
This explains why non-motor features (anosmia, constipation, RBD) precede motor symptoms by years to decades. The model has been challenged due to inconsistencies (e.g., not all patients follow the pattern, asymmetry of clinical findings is not reflected in bilateral pathology), but it remains a useful conceptual framework. - Katzung's Basic and Clinical Pharmacology; Bradley and Daroff's Neurology
6. Prion-Like Propagation ("Body-First" vs "Brain-First" Hypothesis)
A landmark discovery: Lewy pathology was found in healthy embryonic dopamine neurons transplanted into the striatum of PD patients years earlier - meaning the misfolded protein had transferred from diseased host cells to healthy grafted neurons.
This led to the prion hypothesis of PD:
- Misfolded α-synuclein can be released from one neuron and taken up by a neighboring cell
- Once internalized, misfolded protein acts as a template, inducing misfolding of native α-synuclein ("permissive templating")
- Pathology spreads in a stereotyped, trans-neuronal pattern consistent with Braak staging
Gut-brain axis: Environmental triggers (possibly pathogens, toxins) in the gut may initiate α-synuclein misfolding in the enteric nervous system (ENS), which then spreads via the vagus nerve to the brainstem - this is the "body-first" hypothesis. Epidemiological data showing reduced PD rates in people who underwent vagotomy offers supporting evidence. - Harrison's Principles of Internal Medicine 22e; Goldman-Cecil Medicine
7. Neuroinflammation
- Activated microglia are found in the SNc of PD patients and in MPTP animal models
- Pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) contribute to dopaminergic neuron death
- α-Synuclein aggregates can act as DAMPs (damage-associated molecular patterns), activating microglial and astrocytic inflammatory responses
- T-cell infiltration has been documented in the SNc of PD patients
- Maladaptive immune responses, possibly initiated in the gut, are now recognized as accelerating PD pathogenesis - per the 2024 Lancet review by Morris et al. (PMID: 38245249)
8. Basal Ganglia Circuit Dysfunction
The net result of SNc dopamine neuron loss is a disruption of direct and indirect basal ganglia pathways:
- Direct pathway (striatum → GPi/SNr, inhibitory): Normally dopamine activates D1 receptors, facilitating movement. With dopamine loss, this pathway is underactive → less inhibition of GPi → more GPi output
- Indirect pathway (striatum → GPe → STN → GPi): Normally dopamine inhibits D2 receptors here, dampening the pathway. With dopamine loss, this pathway is overactive → more STN excitation of GPi → more GPi output
- Net result: Excessive inhibitory output from GPi/SNr → over-suppression of thalamocortical circuits → hypokinesia, bradykinesia, rigidity
This model predicts that lesioning or stimulating the STN or GPi (deep brain stimulation targets) would restore movement - which is borne out clinically. - Harrison's Principles of Internal Medicine 22e; Katzung's Basic and Clinical Pharmacology
9. Key Genetic Causes Summary
| Gene / Locus | Inheritance | Age of Onset | Lewy Bodies | Key Mechanism |
|---|---|---|---|---|
| SNCA (PARK1/4) | AD | 30-50s | Yes | α-Synuclein overproduction/aggregation |
| LRRK2 (PARK8) | AD | 50-60s (variable) | Variable | Gain-of-function kinase activity; impaired lysosomal/vesicular trafficking; Rab phosphorylation |
| VPS35 (PARK17) | AD | ~50s | Yes | Retromer complex dysfunction; impaired vesicular trafficking |
| RAB32 | AD | Typical PD range | - | Interacts with LRRK2; newest identified form |
| Parkin (PARK2) | AR | <40 yrs (juvenile) | Rare (~20%) | Impaired ubiquitin E3 ligase; failed UPS; mitophagy defect |
| PINK1 (PARK6) | AR | 20-40s | Yes | Impaired mitophagy; defective mitochondrial quality control |
| DJ-1 (PARK7) | AR | 20-40s | Yes | Impaired oxidative stress response; α-synuclein aggregation |
| GBA1 | Risk factor | Typical onset | Yes | Lysosomal glucocerebrosidase deficiency; ALP impairment; α-synuclein accumulation |
Only ~5-10% of cases are monogenic; the vast majority are multifactorial sporadic disease. - Adams and Victor's Principles of Neurology (12th ed.); Harrison's Principles of Internal Medicine 22e
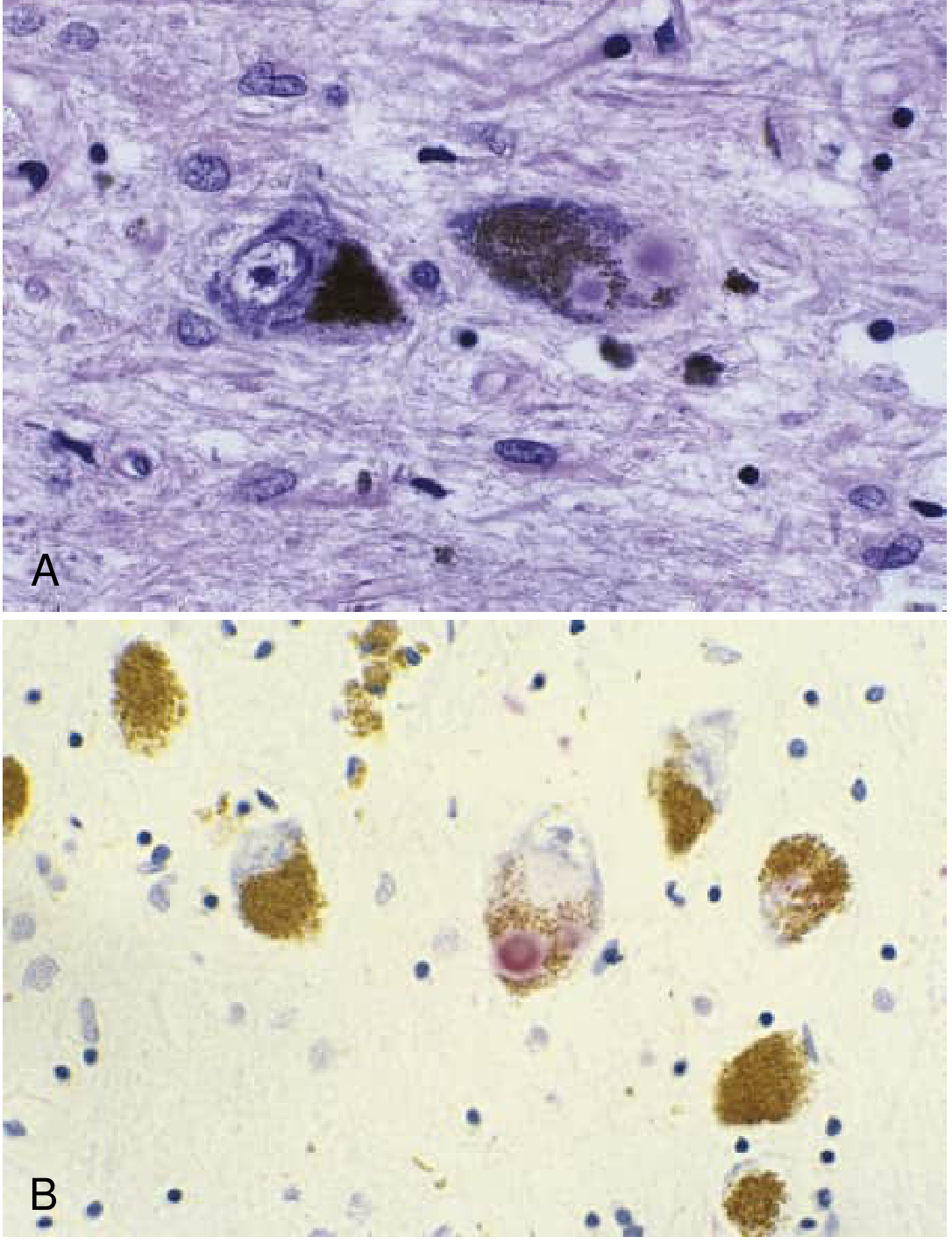
Recent evidence note: A 2024 Lancet review (Morris et al., PMID 38245249) emphasizes the convergence of mitochondrial, lysosomal/endosomal, and immune/inflammatory dysfunction in both genetic and sporadic PD, and highlights the gut-brain axis as a promising pathway for early disease initiation. No disease-modifying therapy has yet demonstrated clinical efficacy, but these mechanistic insights are driving multiple neuroprotective drug programs (GCase enhancers, LRRK2 kinase inhibitors, anti-α-synuclein immunotherapies, c-Abl inhibitors).
Sources: Adams and Victor's Principles of Neurology (12th ed.); Goldman-Cecil Medicine (International Edition); Harrison's Principles of Internal Medicine 22e; Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Bradley and Daroff's Neurology in Clinical Practice; Katzung's Basic and Clinical Pharmacology (16th ed.); Morris et al., Lancet 2024 (PMID 38245249)
This is a shared conversation. Sign in to Orris to start your own chat.