Meckel gruber sybdrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Meckel-Gruber syndrome"[MeSH Terms]
Reading File
Reading File
Reading File
Finding Sources
Reading File
Searching PubMed
"Meckel-Gruber syndrome" AND ciliopathy
Reading File
Reading File
Reading File
Searching the Web
Meckel-Gruber syndrome clinical features genetics diagnosis management 2024
I now have comprehensive information to write a thorough answer. Let me compile everything.
Meckel-Gruber Syndrome (MKS)
Meckel-Gruber syndrome is a lethal autosomal recessive ciliopathy - the most severe end of the ciliopathy spectrum - characterized by a classic triad of malformations affecting the kidneys, central nervous system, and digits.
Classic Diagnostic Triad
| Feature | Description |
|---|---|
| Occipital encephalocele | CNS defect, most commonly posterior (occipital); Dandy-Walker malformation and hydrocephalus also occur |
| Bilateral polycystic kidneys | Cystic renal dysplasia, often massively enlarged; leads to oligohydramnios |
| Postaxial polydactyly | Extra digits on the ulnar/fibular side of hands or feet |
All three components of the triad need not be present simultaneously - incomplete forms ("formes frustes") occur. At minimum, two of the three features are typically required for diagnosis.
Additional Features (Phenotypic Spectrum)
The phenotype is highly variable due to the underlying ciliopathy mechanism:
- Hepatic: Biliary ductal dysgenesis, congenital hepatic fibrosis (CHF), biliary duct proliferation
- Pulmonary: Pulmonary hypoplasia (the leading cause of death) - secondary to oligohydramnios from renal failure
- Craniofacial: Microcephaly, microphthalmia, cleft lip/palate, sloping forehead
- Genital: Ambiguous genitalia, cryptorchidism
- Cardiac: Atrial septal defect, coarctation of the aorta, pulmonary stenosis
- Skeletal: Shortening of long bones, dysplasia
- Other CNS: Arnold-Chiari malformation, absent corpus callosum, cerebellar hypoplasia
Pathophysiology - Ciliopathy Basis
MKS is caused by dysfunction of the primary cilium - a non-motile, antenna-like organelle present on virtually every mammalian cell that coordinates developmental signaling (Hedgehog pathway, Wnt pathway, PDGF signaling).
The causative proteins in MKS localize to the transition zone (TZ) - the gating compartment at the base of the cilium that regulates protein trafficking in and out of the ciliary compartment. Loss of TZ function disrupts multiple developmental pathways simultaneously, explaining the multi-organ phenotype.
The MKS proteins include meckelin (MKS3/TMEM67), which controls basal body positioning and epithelial branching morphogenesis, partly via the non-canonical Wnt pathway.
Genetics
- Inheritance: Autosomal recessive; recurrence risk 25% with each pregnancy
- Incidence: ~1 in 135,000 live births worldwide; much higher in consanguineous populations (e.g., 1 in 500 live births in Qatar; elevated among Gujarati Indians, Tatars, Hutterites, and Finns)
- Extreme genetic heterogeneity: At least 13 genes confirmed (MKS1-MKS13):
| Gene | Locus | Protein |
|---|---|---|
| MKS1 | 17q22 | MKS1 |
| MKS2/TMEM216 | 11q13.1 | Transmembrane protein 216 |
| MKS3/TMEM67 | 8q22.1 | Meckelin |
| MKS4/CEP290 | 12q21.32 | Centrosomal protein 290 |
| MKS5/RPGRIP1L | 16q12.2 | RPGRIP1-like |
| MKS6/CC2D2A | 4p15.32 | CC2 domain protein |
| MKS7/NPHP3 | 3q22.1 | Nephrocystin-3 |
| MKS8/TCTN2 | 12q24.31 | Tectonic-2 |
| MKS9/B9D1 | 17p11.2 | B9 domain containing 1 |
| MKS10/B9D2 | 19q13.2 | B9 domain containing 2 |
| MKS11/TMEM231 | 16q23.1 | Transmembrane protein 231 |
| MKS12/KIF14 | 1q32.1 | Kinesin family member 14 |
| MKS13/TMEM107 | 17p13.1 | Transmembrane protein 107 |
Additional genes include TXNDC15, CEP55, CSPP1, C5orf42, and TMEM17 (described as recently as 2025).
Key genetics note: Multiple MKS genes are allelic with Joubert syndrome (JBTS) and nephronophthisis (NPHP) genes - the same gene can cause MKS in one family and the milder JBTS in another, depending on the specific mutation. This is the "MKS-JBTS-NPHP spectrum."
Prenatal Diagnosis
Diagnosis is typically made prenatally by ultrasound, often as early as 10-14 weeks gestation (first trimester):
- Large echogenic kidneys
- Occipital encephalocele
- Oligohydramnios (due to renal failure)
- Polydactyly
MRI can supplement ultrasound for clearer delineation of CNS anomalies. Confirmatory molecular genetic testing (multi-gene panel) can be performed on amniocentesis or chorionic villus sampling specimens.
In over 75% of prenatally diagnosed cases in European series, therapeutic termination of pregnancy was chosen.
Prognosis
MKS is essentially uniformly lethal when fully expressed:
- Most affected fetuses are stillborn or die within hours to days of birth
- Death is primarily due to pulmonary hypoplasia from severe oligohydramnios
- Rare survivors with incomplete forms have been described; the longest reported survival in a neurosurgically managed cohort was 11 months (median 6 months) - [systematic review 2024, PMID 38459147]
- No curative treatment exists
Management
Management is supportive and family-centered:
- Genetic counseling - recurrence risk 25%; offer carrier testing to parents and siblings
- Prenatal diagnosis - first-trimester ultrasound in subsequent pregnancies; molecular testing if causative mutation identified
- Preimplantation genetic testing (PGT) - now available when the family mutation is known
- Postnatal (if live birth): palliative care focused on comfort; respiratory support
- If neurosurgery considered: repair of encephalocele has been attempted in rare cases; shunting of hydrocephalus may provide short-term benefit, but overall prognosis remains extremely poor
Differential Diagnosis
| Condition | Distinguishing Features |
|---|---|
| Joubert syndrome | Cerebellar vermis aplasia ("molar tooth sign" on MRI), not lethal, similar genetics |
| Nephronophthisis | Progressive tubular disease, typically presents in childhood, less severe |
| Trisomy 13 | Holoprosencephaly, midline defects; chromosomal (not single-gene) |
| ARPKD | Bilateral cystic kidneys but no encephalocele; mutation in PKHD1 |
| Bardet-Biedl syndrome | Retinal dystrophy, obesity, cognitive impairment - milder |
Recent Research (2024-2025)
- A 2025 study in Nature Communications (PMID: 40436881) demonstrated that the MKS protein TMEM67 is cleaved by ADAMTS9, uncoupling Wnt signaling from ciliogenesis - a new mechanistic insight that may explain the pleiotropic phenotype.
- TMEM17 was identified as a new causative gene in 2025 (Clin Genet, PMID not yet indexed), expanding the known gene list.
- Multi-gene panels have dramatically improved molecular diagnosis rates, enabling preimplantation genetic testing (PGT) for families with known mutations.
Sources:
- Brenner and Rector's The Kidney, 2-Volume Set (Cystic Kidney Diseases section)
- Comprehensive Clinical Nephrology, 7th Edition (Table 47.1 - Features of Kidney Cystic Disease)
- Creasy & Resnik's Maternal-Fetal Medicine (Fetal Anomalies section)
- Hartill V et al. Meckel-Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front Pediatr. 2017;5:244
- Junior JHMF et al. Neurosurgical intervention for the Meckel-Gruber Syndrome: A systematic review. Childs Nerv Syst. 2024 Jul. [PMID: 38459147]
Fibroscan
Finding Sources
Finding Sources
Searching PubMed
"transient elastography" AND "liver fibrosis" AND diagnosis
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Searching PubMed
"transient elastography" AND "NAFLD" AND "liver fibrosis"
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive answer. Here it is:
FibroScan (Vibration-Controlled Transient Elastography - VCTE)
What Is FibroScan?
FibroScan is the trade name for vibration-controlled transient elastography (VCTE) - the most widely used noninvasive bedside tool for assessing hepatic fibrosis by measuring liver stiffness. It was FDA-approved in 2013 for use in patients with liver disease.
Principle of Operation
The underlying physics rests on two facts:
- Fibrosis increases the stiffness of hepatic tissue
- A shear wave propagates faster through stiffer material than through elastic material
How it works:
- The ultrasound transducer emits a low-frequency (50 Hz) elastic shear wave through the liver
- The speed of propagation of this wave through a tissue window of 1 cm × 4 cm (100 times the area of an average biopsy specimen) is measured
- Liver stiffness is expressed in kilopascals (kPa)
- Higher kPa = stiffer liver = more fibrosis
The large sampling area is a major advantage over liver biopsy, which only assesses 1/50,000 of total liver volume.
kPa Thresholds by Disease
General / Summary Table (Harrison's 2025)
| Stage | Transient Elastography (kPa) |
|---|---|
| Advanced fibrosis (F3) | >7.3 kPa |
| Cirrhosis (F4) | >15 kPa (range 9-26.5 kPa) |
Hepatitis C
| Finding | Cutoff | LR+ | LR- |
|---|---|---|---|
| Advanced fibrosis | >9.5 kPa | 8.1 | 0.30 |
Hepatitis B
| Finding | Cutoff | LR+ | LR- |
|---|---|---|---|
| Rule out advanced fibrosis | <8.1 kPa | - | 0.16 |
| Advanced fibrosis | >10.5 kPa | 14.4 | 0.29 |
NAFLD / MASLD
| Risk Level | VCTE (kPa) |
|---|---|
| Low (rule out advanced fibrosis) | <8 kPa |
| Intermediate (possible fibrotic MASH) | 8-12 kPa |
| High (likely advanced fibrosis) | >12 kPa |
Alcohol-Associated Liver Disease
| Finding | kPa |
|---|---|
| Normal liver | <6 kPa |
| Advanced fibrosis (≥F3) | >8 kPa |
| Cirrhosis (F4) | >12.5 kPa |
Primary Sclerosing Cholangitis (PSC)
| Fibrosis Stage | kPa Cutoff |
|---|---|
| ≥F1 | 7.4 kPa |
| ≥F2 | 8.6 kPa |
| ≥F3 | 9.6 kPa |
| F4 (cirrhosis) | 14.4 kPa |
Performance
- Cirrhosis detection: AUROC = 0.94 (meta-analysis; excellent)
- Performs best at distinguishing cirrhosis vs. no cirrhosis
- Less accurate for discriminating intermediate fibrosis stages (F1-F2)
- Combining VCTE with serum markers (FibroTest/FIB-4) increases accuracy and can avoid liver biopsy in many patients
Clinical Indications
FibroScan is validated and used in:
- Chronic hepatitis C (staging pre/post-treatment)
- Chronic hepatitis B
- NAFLD/MASLD - now central to all major guidelines (EASL 2024, AASLD 2025)
- Alcohol-associated liver disease
- Primary biliary cholangitis (PBC)
- Hemochromatosis
- Recurrent hepatitis after liver transplantation
- Monitoring hepatotoxicity in patients on methotrexate (e.g., psoriasis)
- PSC - fibrosis staging superior to AST/platelet ratio, FIB-4, and Mayo risk score
Role in MASLD/NAFLD Algorithm (Current Guidelines)
The 2024 EASL-EASD-EASO guidelines recommend a sequential approach:
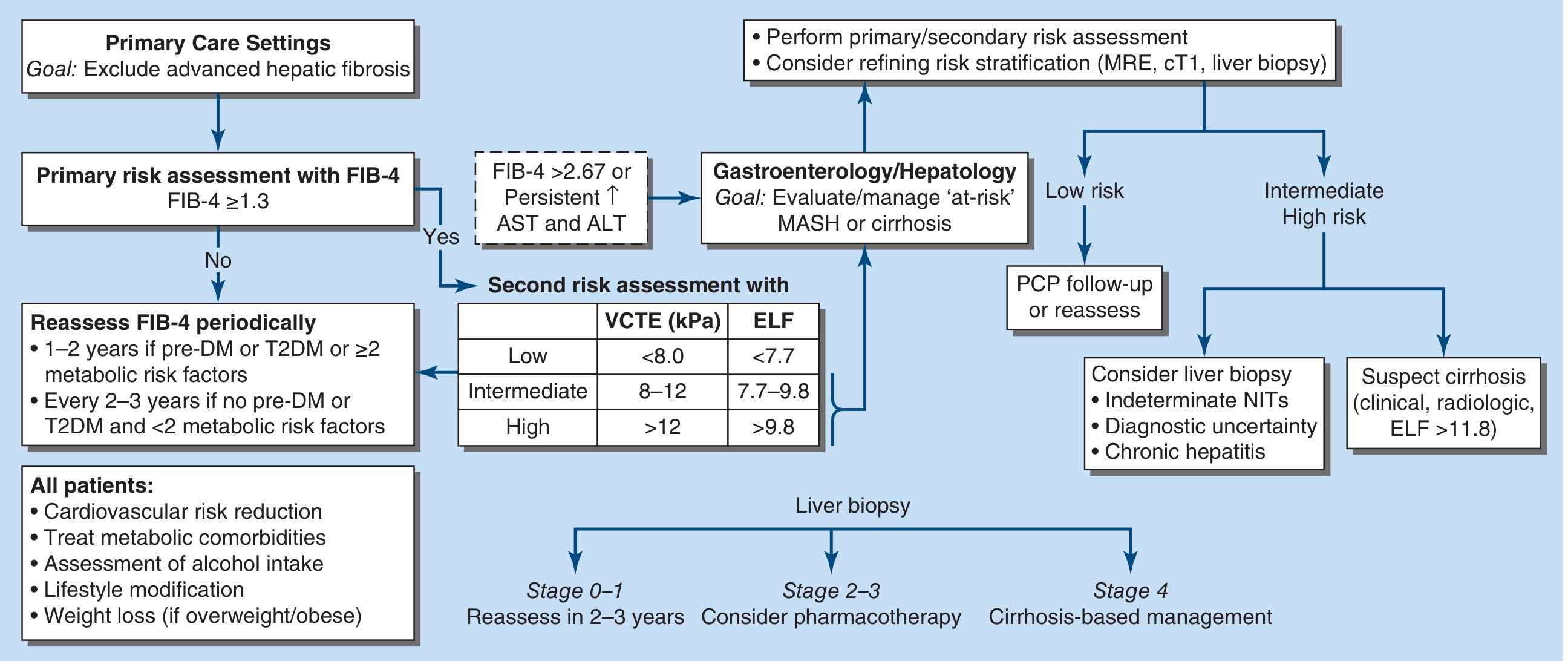
Algorithm for fibrosis risk stratification in MASLD - Harrison's Principles of Internal Medicine 22E (2025)
Step 1: FIB-4 score as primary screening
- FIB-4 <1.3 → low risk → reassess every 1-3 years
- FIB-4 ≥1.3 or >2.67 → proceed to secondary NIT
Step 2 (secondary): VCTE/FibroScan
- LSM <8 kPa → rules out advanced fibrosis
- LSM 8-12 kPa → intermediate; may need specialist referral
- LSM >12 kPa → high risk; consider liver biopsy / specialist management
Limitations & Confounders
FibroScan measures liver stiffness, not fibrosis per se. Stiffness can be elevated by factors other than fibrosis, reducing specificity:
| Confounder | Effect |
|---|---|
| Active hepatic inflammation | Falsely elevated kPa |
| Hepatic edema or congestion | Falsely elevated kPa |
| Hepatocyte necrosis | Falsely elevated kPa |
| Obesity (BMI >30) | Reduced reliability; XL probe needed |
| Intrasinusoidal cellularity (malignant, inflammatory, sickled cells) | Falsely elevated kPa |
| Postprandial state (non-fasting) | Falsely elevated kPa |
| Obstructive jaundice | Falsely elevated kPa |
| Hepatic iron overload | Reduces specificity |
Other technical limitations:
- Cannot assess degree of hepatic inflammation (only stiffness)
- Less reliable for intermediate fibrosis staging (F1-F2)
- Requires patient cooperation (breath-holding)
- 10 valid measurements needed; IQR/median ratio <30% for reliable result
- A positive result (high LSM) has low positive predictive value for cirrhosis (0.34-0.71)
- Does not replace biopsy when inflammation/etiology is uncertain
FibroScan vs. Competing Noninvasive Tests
| Test | Parameters | Advanced Fibrosis Cutoff | Cirrhosis Cutoff |
|---|---|---|---|
| VCTE (FibroScan) | Shear wave speed | >7.3 kPa | >15 kPa |
| ARFI/pSWE | Acoustic radiation force impulse | ~2.0 m/s | ~2.5 m/s |
| MR Elastography | 65 Hz waves (MRI) | ~3.5 kPa | ~5 kPa |
| FIB-4 | Age, AST, ALT, platelets | >1.45 | >3.25 |
| FibroTest/FibroSure | α₂-macroglobulin, haptoglobin, ApoA1, GGT, bilirubin | >0.45 | >0.63 |
| APRI | AST, platelets | >1.0 | >1.5 |
| ELF panel | Hyaluronic acid, TIMP-1, PIIINP | >7.7 | >9.3 |
MR Elastography is more accurate than VCTE for staging fibrosis across all stages but is more expensive and less accessible. VCTE remains the first-line imaging-based tool.
Additional Features of Modern FibroScan Devices
- Controlled Attenuation Parameter (CAP): Measures hepatic steatosis simultaneously (in dB/m). Provides semi-quantitative assessment of fat content at the same examination.
- CAP >248 dB/m = S1 steatosis; >268 = S2; >280 = S3 (severe)
Sources:
- Harrison's Principles of Internal Medicine, 22nd Edition (2025) - Table 347-5, Figure 354-2
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease - Chapters 18, 20, 74, 80
- Symptom to Diagnosis, 4th Edition - Table 17-3
- EASL-EASD-EASO Clinical Practice Guidelines on MASLD 2024 [PMID: 38851997]
- Duarte-Rojo A et al. Imaging-based noninvasive liver disease assessment - AASLD Practice Guideline. Hepatology 2025 [PMID: 38489521]
This is a shared conversation. Sign in to Orris to start your own chat.