Optic atrophy
Reading File
Finding Sources
Searching PubMed
"optic atrophy" OR "optic neuropathy"
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Finding Sources
I now have comprehensive information. Let me compile the full answer.
Optic Atrophy
Optic atrophy is the end-stage morphological result of axonal degeneration in the optic nerve pathway, anywhere from the retrolaminar optic nerve to the lateral geniculate body. It manifests as disc pallor combined with disturbances in visual function.
Definition & Pathophysiology
Optic atrophy represents late-stage changes following loss of retinal ganglion cell axons. Axon loss leads to:
- Replacement of normal neural tissue by fibrous (glial) tissue
- Reduction in disc vascularity
- Thinning of the retinal nerve fibre layer (RNFL)
- Pallor of the optic disc
Notably, optic atrophy from acquired postgeniculate lesions (posterior to the lateral geniculate nucleus) does NOT produce disc pallor - however, congenital postgeniculate lesions can cause pallor via transsynaptic degeneration.
Classification
1. Primary Optic Atrophy
Occurs without antecedent disc swelling. Results from lesions affecting the visual pathway from the retrolaminar optic nerve to the lateral geniculate body.
Fundus appearance: Flat, chalk-white disc with sharply delineated margins; reduced number of disc surface vessels; attenuated peripapillary vasculature; RNFL thinning.
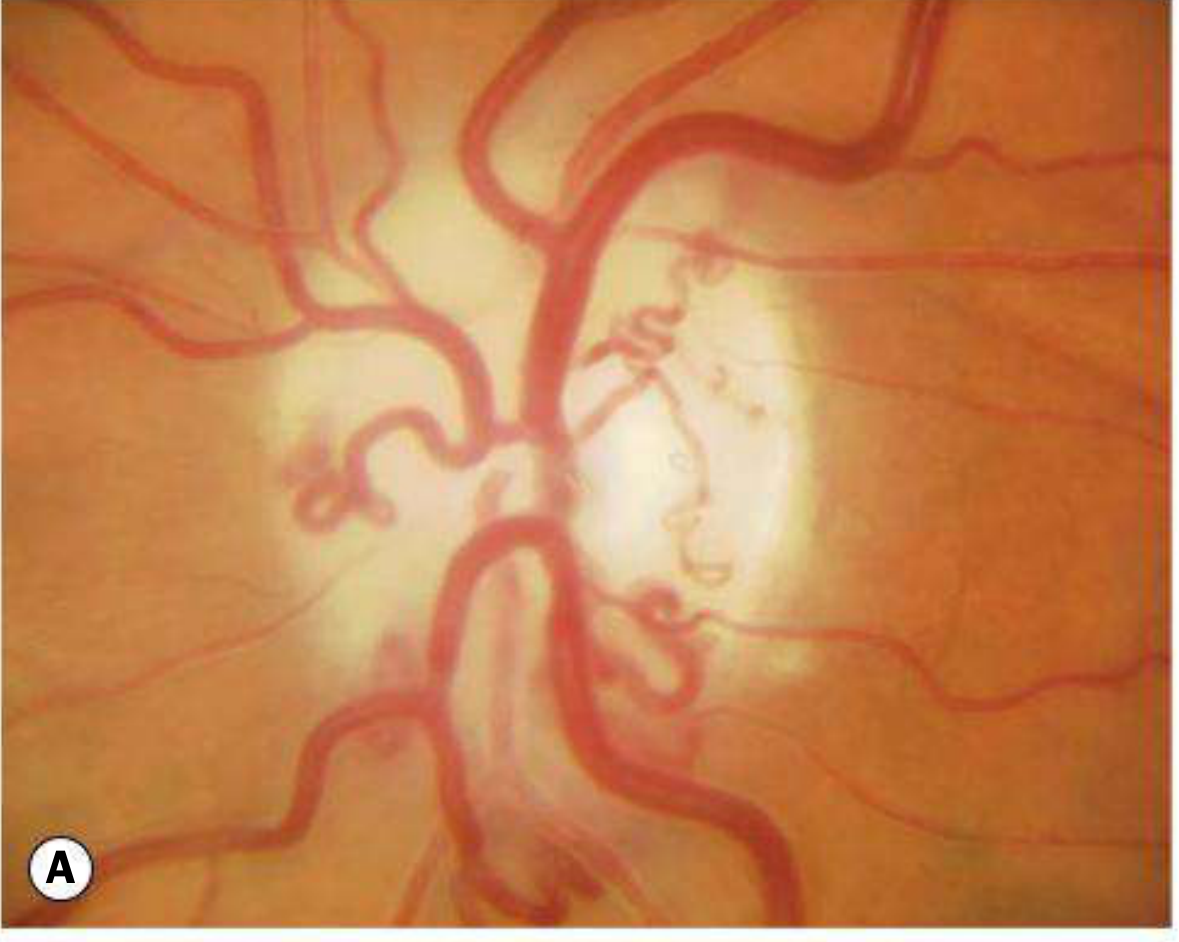
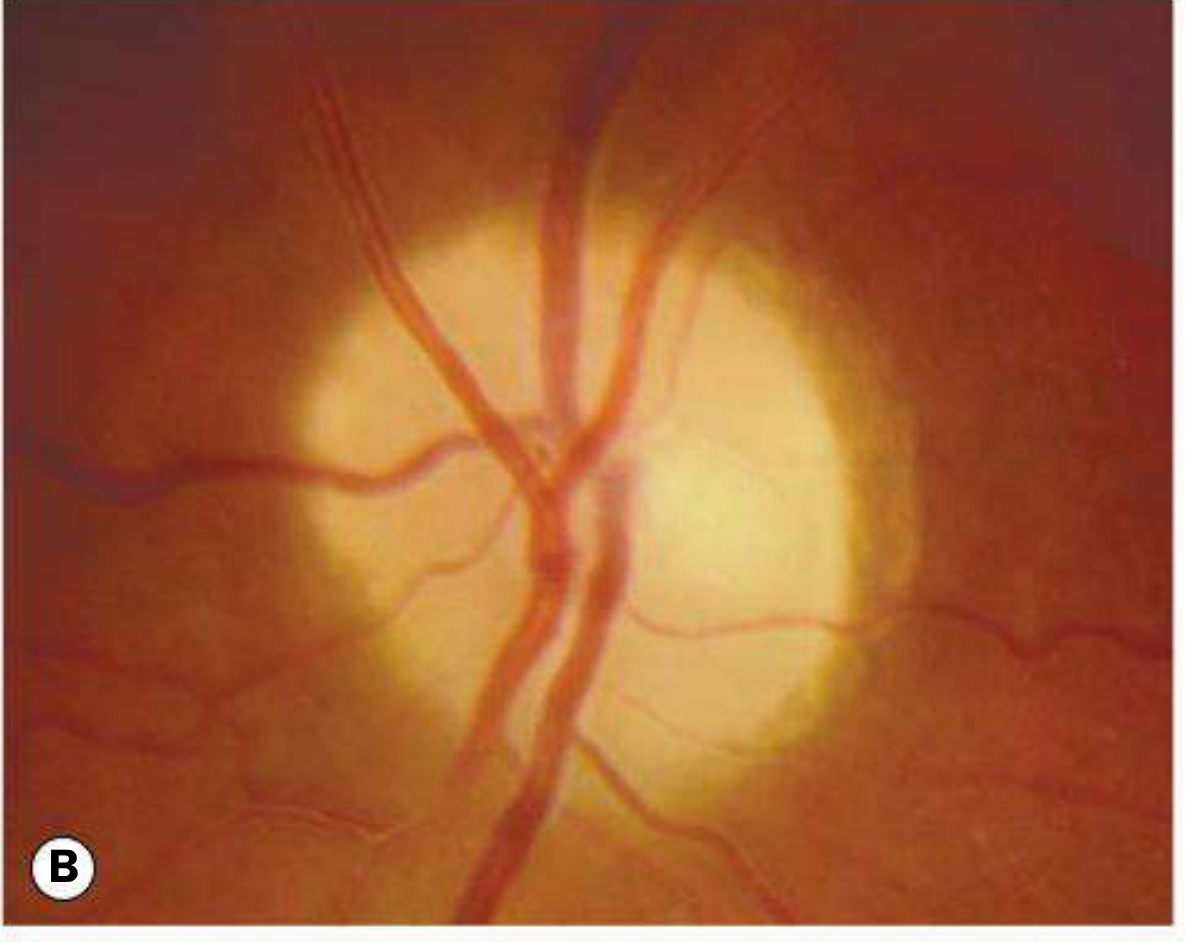
Patterns of pallor:
- Temporal pallor - indicates atrophy of the papillomacular bundle; classically seen after demyelinating optic neuritis, and in toxic/nutritional optic neuropathy
- Band (bow-tie) atrophy - nasal and temporal pallor from loss of fibres entering nasally and temporally; seen with chiasmal or optic tract lesions
Level of lesion:
- Anterior to chiasm → unilateral atrophy
- At chiasm or optic tract → bilateral changes
Important causes:
- Optic neuritis (demyelinating > other)
- Compression by tumours or aneurysms
- Hereditary optic neuropathies (e.g., LHON, dominant optic atrophy)
- Toxic/nutritional optic neuropathy
- Trauma
2. Secondary Optic Atrophy
Preceded by longstanding swelling (papilloedema or papillitis) of the optic nerve head.
Fundus appearance: Slightly or moderately raised disc, white or greyish, with poorly delineated margins due to gliosis; obscuration of the lamina cribrosa; reduced disc vessels; Paton lines (peripapillary circumferential retinochoroidal folds) may persist; arteriolar sheathing and venous tortuosity may be present.
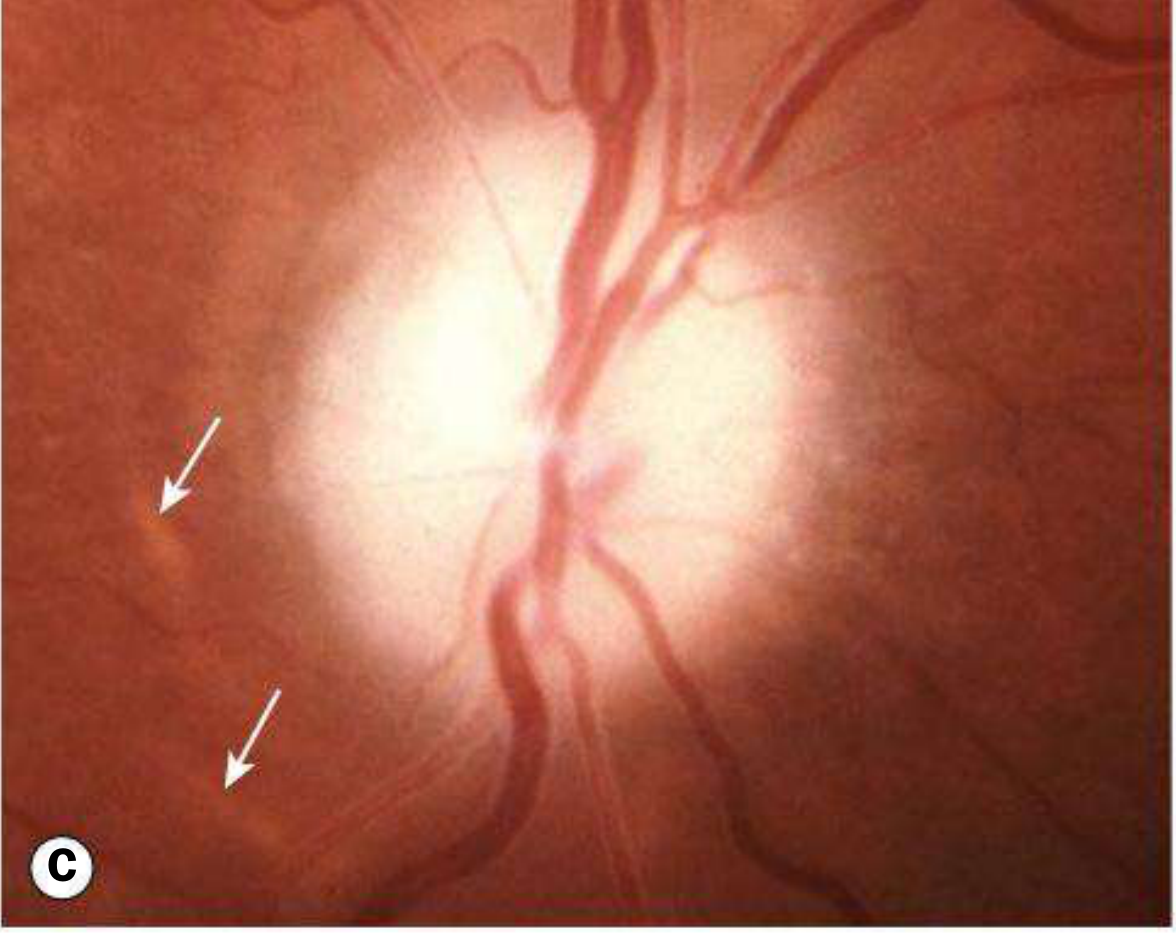
Causes: Chronic papilloedema, anterior ischaemic optic neuropathy, papillitis.
Key distinguishing feature from primary: Margins are blurred/indistinct (primary = sharp).
3. Consecutive Optic Atrophy
Caused by disease of the inner retina or its blood supply - not a primary optic nerve disease.
Fundus appearance: Waxy disc with reasonably preserved architecture.
Causes: Extensive retinal photocoagulation, retinitis pigmentosa, prior central retinal artery occlusion.
4. Glaucomatous Optic Atrophy
Characterised by optic disc cupping (vertical C:D ratio enlargement) WITHOUT disc pallor in typical cases - the key distinguishing feature from other atrophies. Most common optic neuropathy overall.
- Normal-tension glaucoma: cupping + progressive field loss despite normal IOP
- Compressive lesions can occasionally mimic glaucoma with cupping - central visual loss and disc pallor favour compression
Hereditary Optic Atrophies
Dominant Optic Atrophy (Kjer type / OPA1)
| Feature | Details |
|---|---|
| Inheritance | Autosomal dominant |
| Incidence | ~1:50,000 (most common hereditary optic neuropathy) |
| Gene | OPA1 on chromosome 3 (present in ~2/3 of pedigrees); other genes possible; X-linked and AR forms exist |
| Mechanism | Mitochondrial dysfunction, membrane instability |
| Presentation | Insidious visual loss in childhood; family history often present |
| Disc findings | Temporal pallor ± temporal excavation; may be subtle; cup enlargement |
| Prognosis | Variable VA (6/12-6/60); very slow progression over decades |
| Systemic | 20% sensorineural hearing loss |
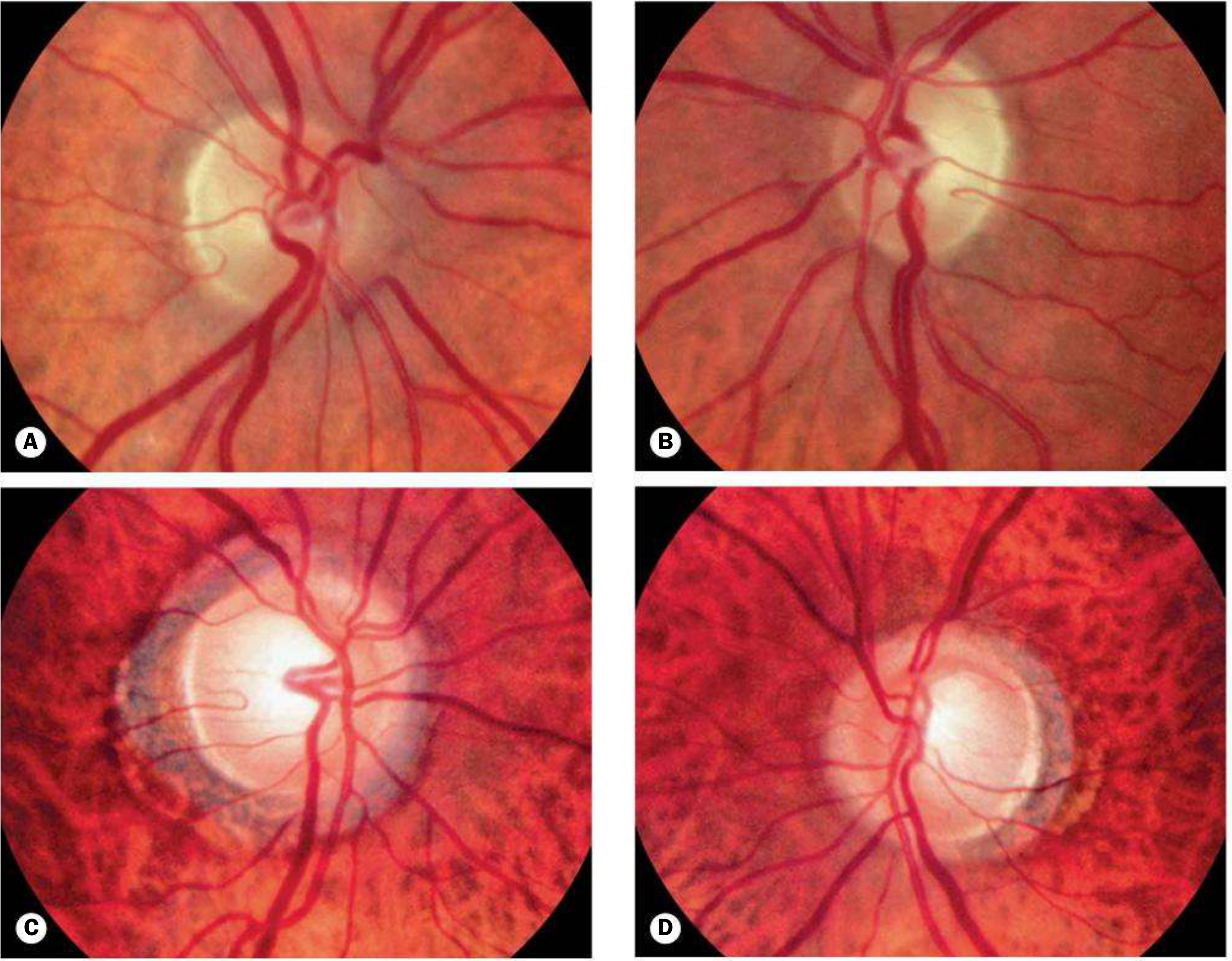
Leber Hereditary Optic Neuropathy (LHON)
- Maternal (mitochondrial) inheritance
- Most common mutations: mt.11778 (ND4), mt.3460 (ND1), mt.14484 (ND6)
- Presentation: acute/subacute painless central visual loss in young males; fellow eye usually affected within weeks to months
- Fundus (acute): peripapillary telangiectatic microangiopathy, disc pseudoedema, RNFL swelling
- Fundus (chronic): optic atrophy develops
- Treatment: idebenone; intravitreal gene therapy (AAV vector expressing normal ND4 cDNA) showing promising results for the 11778 mutation
Wolfram Syndrome (DIDMOAD)
- Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, Deafness
- Genes: WFS1 (Wolfram 1), CISD2 (Wolfram 2), mitochondrial
- Presentation: age 5-21 years; diabetes mellitus typically first
- Disc: diffuse, severe atrophy ± cupping
- Prognosis: poor (final VA < 6/60); reduced life expectancy
- Additional features: anosmia, ataxia, seizures, endocrine abnormalities
Behr Syndrome
- Autosomal recessive; presents in early childhood
- Diffuse optic atrophy + spastic gait, ataxia, intellectual disability
Causes Summary (by Category)
| Category | Examples |
|---|---|
| Inflammatory / demyelinating | Optic neuritis (MS), NMO/NMOSD |
| Ischaemic | AION (arteritic or non-arteritic), CRAO |
| Compressive | Pituitary tumour, meningioma, aneurysm, optic glioma |
| Traumatic | Traumatic optic neuropathy |
| Hereditary | Dominant OA (OPA1), LHON, Wolfram, Behr |
| Glaucomatous | Open-angle, angle-closure, normal-tension |
| Toxic / nutritional | Methanol, ethambutol, B12 deficiency, tobacco-alcohol amblyopia |
| Infective | Syphilis, TB |
| Retinal | Retinitis pigmentosa, CRAO (consecutive) |
| Infiltrative / storage | Tay-Sachs, other lysosomal storage diseases |
| Iatrogenic | Radiation optic neuropathy, extensive photocoagulation |
Investigations
Directed at identifying the underlying cause:
- Visual acuity and colour vision (Ishihara/Hardy-Rand-Rittler) - colour defect often disproportionate in optic nerve disease
- Pupillary assessment - relative afferent pupillary defect (RAPD) in unilateral or asymmetric cases
- Visual field testing (perimetry) - pattern indicates level of lesion
- OCT - RNFL thinning quantified; macular ganglion cell complex
- VEP (visual evoked potential) - delayed and/or reduced amplitude
- Neuroimaging - MRI orbits/brain (with gadolinium) to exclude compressive or infiltrative lesions; mandatory for unexplained or progressive atrophy
- Blood tests - FBC, ESR/CRP, B12, folate, syphilis serology, autoimmune screen (ANA, ANCA, NMO-IgG), genetic testing for OPA1/mitochondrial mutations
- Mitochondrial DNA analysis - if LHON suspected
Management Principles
Treatment is directed at the underlying cause where possible:
| Cause | Intervention |
|---|---|
| Demyelinating optic neuritis | IV methylprednisolone (hastens recovery; long-term VA similar) |
| Compressive | Surgical decompression |
| Nutritional/toxic | Remove toxin; B12 supplementation |
| Glaucoma | IOP reduction (drops, laser, surgery) |
| LHON | Idebenone; gene therapy (ND4 AAV) |
| Ischaemic (arteritic) | High-dose steroids immediately (GCA) |
| Traumatic | Systemic steroids ± optic canal decompression |
Important: Established optic atrophy with axonal loss is largely irreversible with current treatments. The goal is to halt or slow progression.
A Note on Recent Evidence
Two recent meta-analyses (2025-2026) have highlighted that GLP-1 receptor agonists (e.g., semaglutide) may be associated with an increased risk of non-arteritic anterior ischaemic optic neuropathy (NAION), which can lead to optic atrophy [PMID: 40810985; PMID: 41498755]. This is an emerging safety signal worth noting in patients on these agents who develop visual symptoms.
Sources: Kanski's Clinical Ophthalmology 10th ed., p. 780-791 | Wills Eye Manual | Bradley and Daroff's Neurology in Clinical Practice
This is a shared conversation. Sign in to Orris to start your own chat.