Pathophysiology of asthma in PG level
Reading File
Finding Sources
Finding Sources
Searching PubMed
"asthma"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Reading File
Reading File
Reading File
Searching Images
asthma pathophysiology airway inflammation diagram
Reading File
Reading File
Reading File
Reading File
I now have comprehensive material from top-tier sources. Let me compile the complete PG-level answer:
Pathophysiology of Asthma
Sources: Robbins & Cotran Pathologic Basis of Disease (10th ed.), Fishman's Pulmonary Diseases and Disorders, Murray & Nadel's Textbook of Respiratory Medicine
Definition & Core Triad
Asthma is a chronic inflammatory disease of the airways characterized by three interacting pathophysiologic components:
- Airway hyperresponsiveness (AHR)
- Variable airflow obstruction (reversible bronchoconstriction)
- Chronic airway inflammation with structural remodeling
The complex interplay between these factors — not any one element alone — defines the clinical expression of disease.
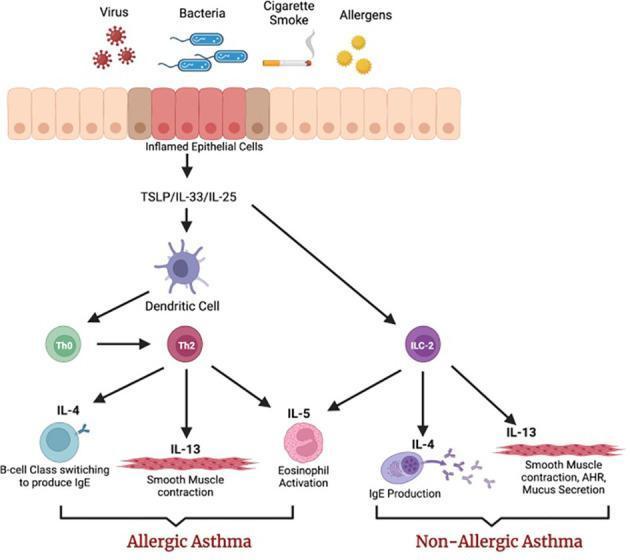
Types of Asthma
| Type | Mechanism |
|---|---|
| Atopic (Allergic) | Th2/IgE-mediated; most common (65–80%) |
| Non-atopic (Intrinsic) | Non-IgE; triggered by cold, exercise, pollutants |
| Drug-induced | ASA/NSAIDs via COX inhibition → ↑ leukotrienes |
| Occupational | Sensitization to workplace antigens |
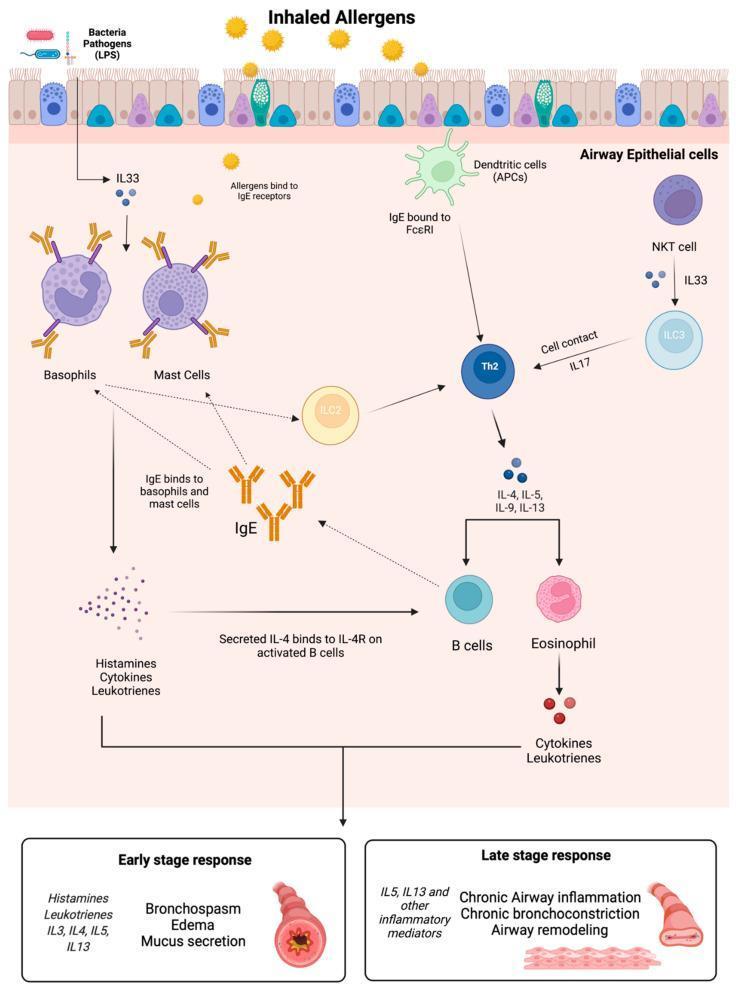
Central Immunopathogenesis: T2 Inflammatory Pathway
Step 1 — Allergen Sensitization (Adaptive Response)
- Inhaled allergen is trapped in the mucus layer and endocytosed by dendritic cells (DCs) whose dendrites interdigitate between bronchial epithelial cells.
- DCs migrate to local lymph nodes, presenting antigen via MHC II to naïve CD4⁺ T cells.
- Under the influence of IL-4 (from mast cells and basophils), naïve T cells differentiate into Th2 cells.
- Th2 cells secrete the critical cytokine triad:
- IL-4 → drives IgE class switching in B cells; promotes AHR in smooth muscle
- IL-5 → eosinophil maturation, recruitment, and activation
- IL-13 → goblet cell mucus hypersecretion; promotes IgE production
- IgE produced by plasma cells binds the high-affinity FcεRI receptors on submucosal mast cells and circulating basophils.
Step 2 — Re-exposure: Mast Cell Activation
On re-exposure, allergen cross-links IgE on mast cells → degranulation.
Preformed mediators released:
- Histamine
- Tryptase, chymase, heparin
- TNF-α, VEGF
Newly synthesized mediators:
- Leukotrienes (LTC4, LTD4, LTE4): potent, prolonged bronchoconstriction + ↑ vascular permeability + ↑ mucus
- PGD2: bronchoconstriction + vasodilation
- Thromboxane A2, PAF
- Cytokines: IL-4, IL-5, IL-8, IL-13, GM-CSF
Biphasic Bronchial Response
Early Phase Reaction (Minutes 0–30)
- Bronchoconstriction: mast cell–released histamine and leukotrienes act directly + via vagal reflexes on subepithelial parasympathetic receptors → airway smooth muscle (ASM) contraction
- Increased vascular permeability → edema
- Mucus hypersecretion (goblet cell stimulation)
- Vasodilation
Late Phase Reaction (Hours 2–8)
- Recruitment of eosinophils, neutrophils, basophils, lymphocytes, monocytes to the airway wall
- Eosinophil-derived:
- Major Basic Protein (MBP), Eosinophil Cationic Protein (ECP) → epithelial damage, ciliary dysfunction
- LTC4 → bronchoconstriction
- Reactive oxygen species (ROS) → oxidative stress
- Charcot-Leyden crystals (crystallized galectin-10/GAL10 from eosinophils) → strong inducers of inflammation and mucus production
- This phase underlies persistent AHR and sets the stage for remodeling
Key Cellular Players
| Cell | Role in Asthma |
|---|---|
| Mast cells | Early-phase trigger; preformed + newly synthesized mediators |
| Th2 / ILC2 cells | Master regulators of T2 inflammation; IL-4, IL-5, IL-13 |
| Eosinophils | Late-phase tissue damage; MBP, ECP, LTC4, ROS |
| Basophils | IgE-dependent histamine + IL-4/IL-13 release; Th2 polarization |
| Dendritic cells | Antigen presentation; Th2 polarization |
| Macrophages | Source of ROS; amplify inflammation |
| Neutrophils | Recruited by LTB4; prominent in severe/steroid-resistant asthma |
| Airway epithelial cells | Release alarmins (TSLP, IL-25, IL-33) after injury |
Innate Immune Contribution: Alarmins & ILC2s
Damaged epithelial cells release epithelial-derived alarmins:
- TSLP (thymic stromal lymphopoietin)
- IL-33
- IL-25
These activate type 2 innate lymphoid cells (ILC2s), which produce IL-4, IL-5, and IL-13 without antigen recognition. This explains non-allergic asthma and explains why inflammation can occur even without adaptive immune sensitization.
Airway Hyperresponsiveness (AHR)
AHR is defined as an exaggerated bronchoconstrictor response to stimuli (cold air, exercise, methacholine, histamine) that would be innocuous in normal individuals.
Mechanisms include:
- Subepithelial exposure of sensory nerves (from epithelial damage) → exaggerated reflex bronchoconstriction
- ASM hypertrophy + hyperplasia → increased contractile mass
- Increased mucosal permeability (allows allergens/irritants deeper access)
- Neural plasticity: upregulation of substance P and other neuropeptides
- Persistent eosinophilic inflammation lowering the threshold of ASM contraction
Mediator Profile Summary
| Mediator | Source | Effect |
|---|---|---|
| Histamine | Mast cells, basophils | Bronchoconstriction, edema |
| LTC4/LTD4/LTE4 | Mast cells, eosinophils | Prolonged bronchoconstriction, mucus, edema |
| LTB4 | Neutrophils | Neutrophil chemotaxis |
| PGD2 | Mast cells | Bronchoconstriction, vasodilation |
| IL-5 | Th2, ILC2 | Eosinophil maturation/activation |
| IL-13 | Th2, ILC2 | Mucus hypersecretion, AHR |
| Acetylcholine | Parasympathetic nerves | ASM contraction via muscarinic receptors |
| MBP / ECP | Eosinophils | Epithelial necrosis, ciliostasis |
| GAL10 (Charcot-Leyden) | Eosinophils | Inflammation, mucus induction |
| TGF-β | Multiple cells | Subepithelial fibrosis (remodeling) |
Genetics & Environment
- Asthma is polygenic — multiple loci on chromosomes 5q, 6p, 11q, 12q, 14q, and 16p are implicated.
- Gene-environment interactions are critical: the same genetic predisposition can result in very different phenotypes depending on:
- Allergen load and type (house dust mite, pollens, cockroach, pet dander)
- Hygiene hypothesis: reduced microbial exposure in early life fails to skew immunity away from Th2, promoting atopy
- Viral URTIs (particularly rhinovirus) → major trigger of exacerbations via TLR3-mediated epithelial alarmin release
Airway Remodeling (Chronic Asthma)
With persistent disease, structural changes accrue and add an irreversible component to obstruction:
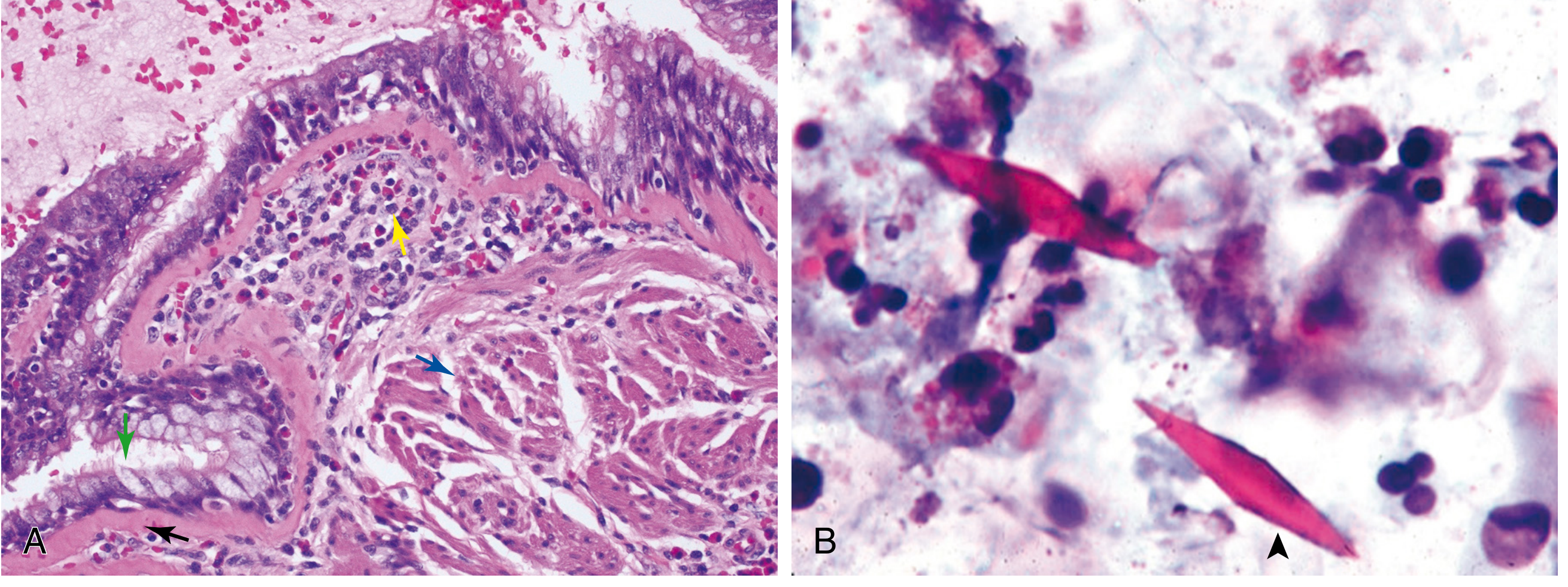
Histology (Robbins Fig. 15.11): (A) Asthmatic bronchus — goblet cell hyperplasia (green arrow), sub-basement membrane fibrosis (black arrow), eosinophilic inflammation (yellow arrow), muscle hypertrophy (blue arrow). (B) Sputum: Charcot-Leyden crystals.
| Structural Change | Mechanism | Consequence |
|---|---|---|
| Goblet cell hyperplasia | IL-13, IL-4 | Mucus plugging |
| Sub-basement membrane fibrosis | TGF-β → collagen deposition | Irreversible narrowing |
| ASM hypertrophy + hyperplasia | Growth factors, chronic contraction | ↑ AHR, fixed obstruction |
| Submucosal gland hypertrophy | Cholinergic + inflammatory stimuli | Mucus hypersecretion |
| Angiogenesis | VEGF from mast cells | Mucosal edema |
| Nerve remodeling | Substance P, VIP changes | Neural AHR |
Growth factors driving remodeling: TGF-β, EGF, FGF-2, VEGF, PDGF.
Aspirin-Exacerbated Respiratory Disease (AERD)
A distinct phenotype with:
- Chronic rhinosinusitis + nasal polyps + severe asthma
- Mechanism: Aspirin/NSAIDs inhibit COX → ↓ PGE2 → loss of tonic inhibition of 5-lipoxygenase → massive surge in cysteinyl leukotrienes (LTC4/D4/E4)
- Mast cells are the primary source of cysteinyl leukotrienes in this syndrome
- Diagnosed by elevated urinary LTE4
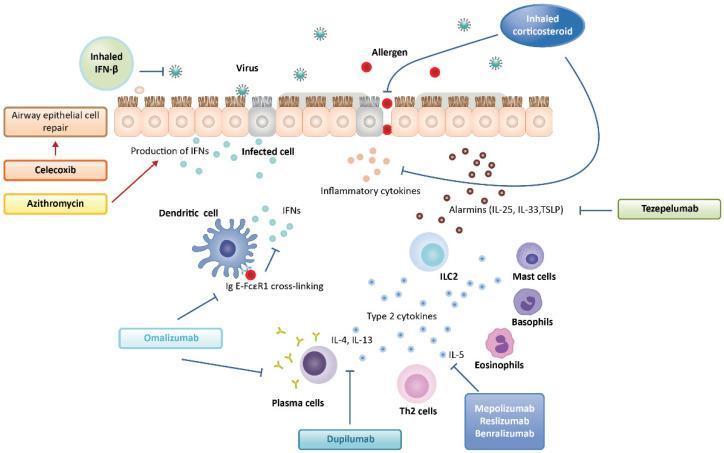
Non-T2 (Th1/Th17) Pathway
In ~20–35% of asthmatic patients (typically severe, steroid-resistant):
- Th17 cells → IL-17 → neutrophil recruitment → neutrophilic inflammation
- Th1 cells → IFN-γ → further epithelial damage
- Often associated with obesity, smoking, pollutant exposure
- Less responsive to inhaled corticosteroids
Physiologic Consequences
- FEV1/FVC ratio < 0.70 (obstructive pattern); reversible with bronchodilator (≥12% + 200 mL improvement)
- Air trapping → ↑ RV, ↑ FRC
- Ventilation-perfusion mismatch → hypoxemia
- Hypocapnia (early, due to hyperventilation); hypercapnia in severe/status asthmaticus (fatigue, impending respiratory failure)
- Pulsus paradoxus > 10 mmHg in severe attacks
- Mucus plugging → atelectasis
Summary Diagram Logic
Allergen → Dendritic cell → Th2 polarization → IgE production → Mast cell sensitization
↓ (Re-exposure)
IgE cross-linking → Mast cell degranulation
↓
Early phase: Bronchoconstriction + edema + mucus (0–30 min)
↓
Late phase: Eosinophil/neutrophil infiltration + epithelial damage (2–8 h)
↓ (Repeated)
Chronic inflammation → Airway remodeling → Fixed obstruction
Key references: Robbins, Cotran & Kumar — Pathologic Basis of Disease, 10th ed., p. 639–641; Fishman's Pulmonary Diseases and Disorders, Ch. 43; Murray & Nadel's Textbook of Respiratory Medicine
Give me a clear flowchart for ppt
Reading File
Reading File
Reading File
Running Command
ls /home/daytona/workspace/ 2>/dev/null || mkdir -p /home/daytona/workspace/asthma-ppt
Running Command
mkdir -p /home/daytona/workspace/asthma-ppt
Writing File
~/asthma-ppt/asthma_flowchart.js
const pptxgen = require("pptxgenjs");
const pres = new pptxgen();
pres.layout = "LAYOUT_WIDE"; // 13.3" x 7.5"
pres.title = "Pathophysiology of Asthma";
// ─── COLOR PALETTE ───────────────────────────────────────────────────────────
const C = {
bg: "0D1B2A", // deep navy background
panel: "112233", // slightly lighter panel
blue1: "1565C0", // sensitization
blue2: "1976D2",
teal: "00838F", // mast cell
orange: "E65100", // early phase
purple: "6A1B9A", // late phase
red: "B71C1C", // remodeling
gold: "F9A825", // accent / arrow
white: "FFFFFF",
lt: "E3EAF4", // light text
dim: "8EA8C3", // dimmed text
};
// helper: draw a rounded box node
function node(slide, x, y, w, h, color, title, sub, titleSize = 11, subSize = 9) {
slide.addShape("roundRect", {
x, y, w, h, rectRadius: 0.12,
fill: { color },
shadow: { type: "outer", color: "000000", blur: 8, offset: 3, angle: 135, opacity: 0.35 }
});
const lines = [];
lines.push({ text: title, options: { bold: true, fontSize: titleSize, color: C.white, breakLine: sub ? true : false } });
if (sub) lines.push({ text: sub, options: { fontSize: subSize, color: "DDDDDD" } });
slide.addText(lines, { x, y, w, h, align: "center", valign: "middle", margin: 4 });
}
// helper: vertical arrow
function arrowDown(slide, cx, y, len, color = C.gold) {
slide.addShape("line", {
x: cx, y, w: 0, h: len,
line: { color, width: 2.5, endArrowType: "triangle" }
});
}
// helper: horizontal arrow (right)
function arrowRight(slide, x, cy, len, color = C.gold) {
slide.addShape("line", {
x, y: cy, w: len, h: 0,
line: { color, width: 2.5, endArrowType: "triangle" }
});
}
// helper: label on arrow
function arrowLabel(slide, x, y, w, text) {
slide.addText(text, { x, y, w, h: 0.3, fontSize: 8, color: C.gold, align: "center", bold: true });
}
// ═══════════════════════════════════════════════════════════════════════════
// SLIDE 1 — TITLE
// ═══════════════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
s.background = { color: C.bg };
// Top accent bar
s.addShape("rect", { x: 0, y: 0, w: 13.3, h: 0.08, fill: { color: C.gold } });
// Main title
s.addText("Pathophysiology of Asthma", {
x: 0.7, y: 1.8, w: 11.9, h: 1.4,
fontSize: 44, bold: true, color: C.white, align: "center",
fontFace: "Calibri"
});
// Subtitle
s.addText("A Systematic Flowchart for PG-Level Understanding", {
x: 0.7, y: 3.3, w: 11.9, h: 0.6,
fontSize: 20, color: C.gold, align: "center", italic: true
});
// Three pillars
const pillars = [
{ x: 1.2, label: "Airway\nHyperresponsiveness", color: C.blue1 },
{ x: 5.15, label: "Variable Airflow\nObstruction", color: C.teal },
{ x: 9.1, label: "Chronic Airway\nInflammation", color: C.purple },
];
pillars.forEach(p => {
s.addShape("roundRect", { x: p.x, y: 4.2, w: 3.0, h: 0.9, rectRadius: 0.12, fill: { color: p.color } });
s.addText(p.label, { x: p.x, y: 4.2, w: 3.0, h: 0.9, fontSize: 12, bold: true, color: C.white, align: "center", valign: "middle" });
});
// Core triad label
s.addText("CORE TRIAD", { x: 0.5, y: 3.9, w: 12.3, h: 0.28, fontSize: 10, color: C.dim, align: "center", bold: true, charSpacing: 4 });
// Bottom bar
s.addShape("rect", { x: 0, y: 7.42, w: 13.3, h: 0.08, fill: { color: C.gold } });
s.addText("Robbins & Cotran | Fishman's Pulmonary | Murray & Nadel", {
x: 0.5, y: 7.1, w: 12.3, h: 0.3, fontSize: 9, color: C.dim, align: "center"
});
}
// ═══════════════════════════════════════════════════════════════════════════
// SLIDE 2 — MASTER FLOWCHART (Main Pathogenesis)
// ═══════════════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
s.background = { color: C.bg };
// Title bar
s.addShape("rect", { x: 0, y: 0, w: 13.3, h: 0.55, fill: { color: C.blue1 } });
s.addText("Asthma Pathogenesis — Master Flowchart", {
x: 0.2, y: 0, w: 12.9, h: 0.55, fontSize: 18, bold: true, color: C.white, valign: "middle"
});
// ── COLUMN LAYOUT (5 rows, center column) ───────────────────────────────
const cx = 5.05; // center x of main column
const nw = 3.2; // node width
const nh = 0.62; // node height
// Row y positions
const rows = [0.75, 1.6, 2.45, 3.3, 4.15, 5.0, 5.85, 6.5];
// ── MAIN CHAIN ──────────────────────────────────────────────────────────
// 1. Trigger
node(s, cx, rows[0], nw, nh, "1A3A5C", "TRIGGER / ALLERGEN", "House dust mite · pollens · pet dander · viruses");
arrowDown(s, cx + nw/2, rows[0] + nh, 0.22);
// 2. Epithelial damage → Alarmins
node(s, cx, rows[1], nw, nh, "1565C0", "EPITHELIAL DAMAGE", "Releases alarmins: TSLP · IL-25 · IL-33");
arrowDown(s, cx + nw/2, rows[1] + nh, 0.22);
// 3. Dendritic cell / ILC2
node(s, cx, rows[2], nw, nh, C.teal, "ANTIGEN PRESENTATION", "Dendritic cells → Th2 polarization | ILC2 activation");
arrowDown(s, cx + nw/2, rows[2] + nh, 0.22);
// 4. Th2 cytokine release
node(s, cx, rows[3], nw, nh, "00695C", "Th2 / ILC2 CYTOKINES", "IL-4 · IL-5 · IL-13 · IL-9");
arrowDown(s, cx + nw/2, rows[3] + nh, 0.22);
// 5. IgE + Mast cell sensitization
node(s, cx, rows[4], nw, nh, C.teal, "IgE PRODUCTION + MAST CELL SENSITIZATION", "B cells → IgE → binds FcεRI on mast cells");
arrowDown(s, cx + nw/2, rows[4] + nh, 0.22);
// 6. Re-exposure: mast cell degranulation
node(s, cx, rows[5], nw, nh, C.orange, "RE-EXPOSURE → MAST CELL DEGRANULATION", "IgE cross-linking → preformed + newly synthesised mediators");
arrowDown(s, cx + nw/2, rows[5] + nh, 0.22);
// 7. Split label
s.addText("BIPHASIC RESPONSE", {
x: cx, y: rows[6] + 0.02, w: nw, h: 0.25,
fontSize: 9, bold: true, color: C.gold, align: "center", charSpacing: 3
});
// ── LEFT BRANCH: Early Phase ─────────────────────────────────────────────
const lx = 1.0;
const bw = 3.4;
// connector from main to left
s.addShape("line", { x: cx, y: rows[6] + 0.28, w: -(cx - lx - bw/2 + bw/2), h: 0, line: { color: C.gold, width: 2 } });
s.addShape("line", { x: lx + bw/2, y: rows[6] + 0.28, w: 0, h: 0.35, line: { color: C.gold, width: 2, endArrowType: "triangle" } });
// Horizontal line at branch level
s.addShape("line", { x: lx + bw/2, y: rows[6] + 0.28, w: cx - (lx + bw/2), h: 0, line: { color: C.gold, width: 2 } });
node(s, lx, rows[6] + 0.63, bw, 0.7, C.orange, "EARLY PHASE (0–30 min)",
"Bronchoconstriction · ↑Mucus\nEdema · Vasodilation", 10, 8.5);
// Mediators box under early phase
s.addShape("roundRect", { x: lx, y: rows[6] + 1.42, w: bw, h: 0.72, rectRadius: 0.1, fill: { color: "3E1A00" } });
s.addText([
{ text: "Mediators: ", options: { bold: true, color: C.gold } },
{ text: "Histamine · LTC4/D4/E4\nPGD2 · TXA2 · Acetylcholine", options: { color: "DDDDDD" } }
], { x: lx, y: rows[6] + 1.42, w: bw, h: 0.72, fontSize: 8.5, align: "center", valign: "middle" });
// ── RIGHT BRANCH: Late Phase ─────────────────────────────────────────────
const rx = 8.9;
s.addShape("line", { x: cx + nw, y: rows[6] + 0.28, w: rx + bw/2 - (cx + nw), h: 0, line: { color: C.purple, width: 2 } });
s.addShape("line", { x: rx + bw/2, y: rows[6] + 0.28, w: 0, h: 0.35, line: { color: C.purple, width: 2, endArrowType: "triangle" } });
node(s, rx, rows[6] + 0.63, bw, 0.7, C.purple, "LATE PHASE (2–8 h)",
"Eosinophils · Neutrophils · T cells\nEpithelial damage · Persistent AHR", 10, 8.5);
s.addShape("roundRect", { x: rx, y: rows[6] + 1.42, w: bw, h: 0.72, rectRadius: 0.1, fill: { color: "1A0033" } });
s.addText([
{ text: "Mediators: ", options: { bold: true, color: "CE93D8" } },
{ text: "MBP · ECP · LTB4\nCharcot-Leyden crystals · ROS", options: { color: "DDDDDD" } }
], { x: rx, y: rows[6] + 1.42, w: bw, h: 0.72, fontSize: 8.5, align: "center", valign: "middle" });
// ── BOTTOM: Remodeling ───────────────────────────────────────────────────
// Arrow from center of both branches down
const remY = rows[6] + 2.3;
const remX = 4.3;
const remW = 4.7;
// converging lines
s.addShape("line", { x: lx + bw/2, y: rows[6] + 2.14, w: 0, h: 0.16, line: { color: C.red, width: 1.5 } });
s.addShape("line", { x: rx + bw/2, y: rows[6] + 2.14, w: 0, h: 0.16, line: { color: C.red, width: 1.5 } });
s.addShape("line", { x: lx + bw/2, y: remY - 0.02, w: rx + bw/2 - (lx + bw/2), h: 0, line: { color: C.red, width: 1.5 } });
s.addShape("line", { x: 6.65, y: remY - 0.02, w: 0, h: 0.25, line: { color: C.red, width: 2.5, endArrowType: "triangle" } });
node(s, remX, remY + 0.23, remW, 0.68, C.red, "AIRWAY REMODELING (Repeated Inflammation)",
"Sub-BM fibrosis · ASM hypertrophy · Goblet hyperplasia · Angiogenesis · Irreversible obstruction", 10, 8);
// Source footer
s.addText("Source: Robbins & Cotran (10e), Fishman's Pulmonary Diseases", {
x: 0.3, y: 7.35, w: 12.7, h: 0.2, fontSize: 7.5, color: C.dim, align: "right"
});
}
// ═══════════════════════════════════════════════════════════════════════════
// SLIDE 3 — Cytokine & Mediator Map
// ═══════════════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
s.background = { color: C.bg };
s.addShape("rect", { x: 0, y: 0, w: 13.3, h: 0.55, fill: { color: C.teal } });
s.addText("Key Cytokines & Mediators in Asthma", {
x: 0.2, y: 0, w: 12.9, h: 0.55, fontSize: 18, bold: true, color: C.white, valign: "middle"
});
// Central hub
s.addShape("ellipse", { x: 5.4, y: 2.6, w: 2.5, h: 1.4, fill: { color: C.teal },
shadow: { type: "outer", color: "000000", blur: 12, offset: 3, angle: 135, opacity: 0.4 }
});
s.addText("Th2 / ILC2\nCYTOKINE\nHUB", { x: 5.4, y: 2.6, w: 2.5, h: 1.4, fontSize: 12, bold: true, color: C.white, align: "center", valign: "middle" });
// Surrounding nodes
const nodes = [
{ x: 0.4, y: 0.75, w: 2.8, h: 1.0, color: C.blue1, title: "IL-4", sub: "IgE class switch\nTh2 differentiation\nASM AHR ↑" },
{ x: 0.4, y: 2.2, w: 2.8, h: 1.0, color: C.blue2, title: "IL-5", sub: "Eosinophil maturation\nrecruitment & activation" },
{ x: 0.4, y: 3.65, w: 2.8, h: 1.0, color: "1A237E", title: "IL-13", sub: "Goblet cell mucus ↑\nIgE production ↑\nAHR ↑" },
{ x: 0.4, y: 5.1, w: 2.8, h: 1.0, color: "004D40", title: "TSLP / IL-33 / IL-25", sub: "Epithelial alarmins\nILC2 activation" },
{ x: 10.1, y: 0.75, w: 2.8, h: 1.0, color: C.orange, title: "Leukotrienes (LTC4/D4/E4)", sub: "Prolonged bronchoconstriction\n↑Mucus · ↑Permeability" },
{ x: 10.1, y: 2.2, w: 2.8, h: 1.0, color: "BF360C", title: "Histamine", sub: "Rapid bronchoconstriction\nVasodilation · Edema" },
{ x: 10.1, y: 3.65, w: 2.8, h: 1.0, color: C.purple, title: "MBP / ECP (Eosinophils)", sub: "Epithelial necrosis\nCiliostasis · ROS" },
{ x: 10.1, y: 5.1, w: 2.8, h: 1.0, color: "4A148C", title: "TGF-β / VEGF / FGF-2", sub: "Airway remodeling\nFibrosis · Angiogenesis" },
];
nodes.forEach(n => {
node(s, n.x, n.y, n.w, n.h, n.color, n.title, n.sub, 11, 8.5);
// connect to hub
const nx_center = n.x + n.w / 2;
const ny_center = n.y + n.h / 2;
const hub_cx = 6.65;
const hub_cy = 3.3;
if (nx_center < hub_cx) {
// left side
s.addShape("line", { x: n.x + n.w, y: ny_center, w: hub_cx - 2.5 - (n.x + n.w), h: ny_center - hub_cy,
line: { color: C.dim, width: 1, dashType: "dash" } });
} else {
s.addShape("line", { x: hub_cx + 2.5, y: hub_cy, w: n.x - (hub_cx + 2.5), h: ny_center - hub_cy,
line: { color: C.dim, width: 1, dashType: "dash" } });
}
});
s.addText("Source: Robbins & Cotran (10e) | Fishman's Pulmonary Diseases", {
x: 0.3, y: 7.35, w: 12.7, h: 0.2, fontSize: 7.5, color: C.dim, align: "right"
});
}
// ═══════════════════════════════════════════════════════════════════════════
// SLIDE 4 — Airway Remodeling Flowchart
// ═══════════════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
s.background = { color: C.bg };
s.addShape("rect", { x: 0, y: 0, w: 13.3, h: 0.55, fill: { color: C.red } });
s.addText("Airway Remodeling — Chronic Asthma", {
x: 0.2, y: 0, w: 12.9, h: 0.55, fontSize: 18, bold: true, color: C.white, valign: "middle"
});
// Trigger node
node(s, 4.9, 0.75, 3.5, 0.65, "3E0000", "REPEATED INFLAMMATION", "Persistent eosinophilic / neutrophilic infiltration");
arrowDown(s, 6.65, 1.4, 0.25);
// Growth factors
node(s, 4.9, 1.65, 3.5, 0.65, "4E1010", "GROWTH FACTORS RELEASED", "TGF-β · VEGF · EGF · FGF-2 · PDGF");
arrowDown(s, 6.65, 2.3, 0.25);
// Five remodeling outcomes in a row
const outcomes = [
{ color: "B71C1C", title: "Sub-BM\nFibrosis", sub: "TGF-β → collagen\ndeposition" },
{ color: "6A1B9A", title: "ASM\nHypertrophy", sub: "↑Contractile mass\n→ fixed AHR" },
{ color: "1565C0", title: "Goblet Cell\nHyperplasia", sub: "IL-13/IL-4\n→ mucus plugging" },
{ color: "00695C", title: "Submucosal\nGland Hypertrophy", sub: "Cholinergic drive\n→ ↑mucus" },
{ color: "E65100", title: "Angiogenesis", sub: "VEGF\n→ mucosal edema" },
];
const startX = 0.35;
const ow = 2.35;
const oh = 1.1;
const gap = 0.27;
const outY = 2.85;
// horizontal bar to outcomes
s.addShape("line", { x: startX + ow/2, y: 2.55, w: (startX + ow/2) + 4*(ow+gap) - (startX + ow/2), h: 0, line: { color: C.red, width: 1.5 } });
outcomes.forEach((o, i) => {
const ox = startX + i * (ow + gap);
node(s, ox, outY, ow, oh, o.color, o.title, o.sub, 10, 8);
s.addShape("line", { x: ox + ow/2, y: 2.55, w: 0, h: 0.3, line: { color: C.red, width: 1.5, endArrowType: "triangle" } });
});
// converging down
const finalY = 4.2;
s.addShape("line", { x: startX + ow/2, y: outY + oh, w: 0, h: 0.18, line: { color: C.gold, width: 1.5 } });
s.addShape("line", { x: startX + ow/2 + 4*(ow+gap), y: outY + oh, w: 0, h: 0.18, line: { color: C.gold, width: 1.5 } });
s.addShape("line", { x: startX + ow/2, y: outY + oh + 0.18, w: 4*(ow+gap), h: 0, line: { color: C.gold, width: 1.5 } });
s.addShape("line", { x: 6.65, y: outY + oh + 0.18, w: 0, h: 0.25, line: { color: C.gold, width: 2, endArrowType: "triangle" } });
// Final outcome
node(s, 4.2, finalY + 0.43, 5.0, 0.7, C.red, "IRREVERSIBLE (FIXED) AIRFLOW OBSTRUCTION",
"FEV1/FVC persistently ↓ · Poor reversibility with bronchodilators · Steroid resistance", 11, 8.5);
// Comparison table
const ty = 5.45;
s.addShape("rect", { x: 0.5, y: ty, w: 12.3, h: 0.35, fill: { color: C.blue1 } });
s.addText("Reversible vs Irreversible Obstruction", { x: 0.5, y: ty, w: 12.3, h: 0.35, fontSize: 11, bold: true, color: C.white, align: "center", valign: "middle" });
const rows2 = [
["Feature", "Reversible (Acute)", "Irreversible (Remodeled)"],
["Bronchodilator response", "≥12% + 200 mL ↑ in FEV1", "< 12% improvement"],
["Dominant pathology", "Bronchoconstriction + mucus", "Fibrosis + muscle hypertrophy"],
["Steroid response", "Good (ICS effective)", "Partial or resistant"],
["Typical phase", "Early / acute attack", "Severe chronic asthma"],
];
const rh = 0.28;
const colW = [3.2, 4.55, 4.55];
const colX = [0.5, 3.7, 8.25];
rows2.forEach((row, ri) => {
const rowY = ty + 0.35 + ri * rh;
const bg = ri === 0 ? "1A2E4A" : (ri % 2 === 0 ? "162035" : "0D1B2A");
s.addShape("rect", { x: 0.5, y: rowY, w: 12.3, h: rh, fill: { color: bg } });
row.forEach((cell, ci) => {
s.addText(cell, {
x: colX[ci] + 0.05, y: rowY, w: colW[ci] - 0.1, h: rh,
fontSize: ri === 0 ? 9 : 8.5,
bold: ri === 0,
color: ri === 0 ? C.gold : C.lt,
valign: "middle"
});
});
// vertical dividers
s.addShape("line", { x: 3.7, y: rowY, w: 0, h: rh, line: { color: C.dim, width: 0.5 } });
s.addShape("line", { x: 8.25, y: rowY, w: 0, h: rh, line: { color: C.dim, width: 0.5 } });
});
s.addText("Source: Robbins & Cotran (10e) | Fishman's Pulmonary Diseases", {
x: 0.3, y: 7.35, w: 12.7, h: 0.2, fontSize: 7.5, color: C.dim, align: "right"
});
}
// ═══════════════════════════════════════════════════════════════════════════
// SLIDE 5 — AHR + Biphasic Response detail
// ═══════════════════════════════════════════════════════════════════════════
{
const s = pres.addSlide();
s.background = { color: C.bg };
s.addShape("rect", { x: 0, y: 0, w: 13.3, h: 0.55, fill: { color: C.orange } });
s.addText("Biphasic Bronchial Response & AHR Mechanisms", {
x: 0.2, y: 0, w: 12.9, h: 0.55, fontSize: 18, bold: true, color: C.white, valign: "middle"
});
// LEFT: Early Phase
s.addShape("rect", { x: 0.3, y: 0.7, w: 5.9, h: 0.38, fill: { color: C.orange } });
s.addText("EARLY PHASE (Minutes 0–30)", { x: 0.3, y: 0.7, w: 5.9, h: 0.38, fontSize: 13, bold: true, color: C.white, align: "center", valign: "middle" });
const earlyItems = [
{ icon: "▶", text: "IgE cross-linking → mast cell degranulation" },
{ icon: "▶", text: "Histamine & LTC4/D4/E4 release" },
{ icon: "▶", text: "Direct ASM contraction" },
{ icon: "▶", text: "Vagal reflex bronchoconstriction (subepithelial receptors)" },
{ icon: "▶", text: "↑ Vascular permeability → mucosal edema" },
{ icon: "▶", text: "Goblet cell activation → mucus hypersecretion" },
];
earlyItems.forEach((item, i) => {
s.addShape("rect", { x: 0.3, y: 1.1 + i * 0.45, w: 5.9, h: 0.4, fill: { color: i % 2 === 0 ? "2A1500" : "1E1000" } });
s.addText(item.icon + " " + item.text, { x: 0.4, y: 1.1 + i * 0.45, w: 5.7, h: 0.4, fontSize: 9.5, color: C.lt, valign: "middle" });
});
// RIGHT: Late Phase
s.addShape("rect", { x: 7.1, y: 0.7, w: 5.9, h: 0.38, fill: { color: C.purple } });
s.addText("LATE PHASE (Hours 2–8)", { x: 7.1, y: 0.7, w: 5.9, h: 0.38, fontSize: 13, bold: true, color: C.white, align: "center", valign: "middle" });
const lateItems = [
{ icon: "▶", text: "Chemokines recruit eosinophils, neutrophils, T cells" },
{ icon: "▶", text: "Eosinophils release MBP, ECP → epithelial damage" },
{ icon: "▶", text: "Charcot-Leyden crystals → further inflammation" },
{ icon: "▶", text: "LTB4 → sustained neutrophil recruitment" },
{ icon: "▶", text: "Persistent AHR (airway hyperresponsiveness)" },
{ icon: "▶", text: "Sets stage for structural remodeling" },
];
lateItems.forEach((item, i) => {
s.addShape("rect", { x: 7.1, y: 1.1 + i * 0.45, w: 5.9, h: 0.4, fill: { color: i % 2 === 0 ? "150025" : "0D0018" } });
s.addText(item.icon + " " + item.text, { x: 7.2, y: 1.1 + i * 0.45, w: 5.7, h: 0.4, fontSize: 9.5, color: C.lt, valign: "middle" });
});
// Divider
s.addShape("line", { x: 6.65, y: 0.7, w: 0, h: 2.8, line: { color: C.gold, width: 1.5, dashType: "dash" } });
s.addText("VS", { x: 6.3, y: 1.95, w: 0.7, h: 0.4, fontSize: 12, bold: true, color: C.gold, align: "center" });
// AHR mechanisms
s.addShape("rect", { x: 0.3, y: 3.85, w: 12.7, h: 0.38, fill: { color: "1A3A1A" } });
s.addText("AIRWAY HYPERRESPONSIVENESS (AHR) — Mechanisms", {
x: 0.3, y: 3.85, w: 12.7, h: 0.38, fontSize: 13, bold: true, color: "A5D6A7", align: "center", valign: "middle"
});
const ahrItems = [
"Exposed subepithelial sensory nerves → exaggerated reflex bronchoconstriction",
"ASM hypertrophy + hyperplasia → ↑ contractile mass",
"↑ Mucosal permeability → deeper allergen penetration",
"Neural plasticity: ↑ Substance P, VIP changes",
"Persistent eosinophilic inflammation lowers ASM threshold",
"Epithelial damage → loss of bronchodilating epithelium-derived factor (EpDRF)",
];
const ahrPerRow = 3;
ahrItems.forEach((item, i) => {
const col = i % ahrPerRow;
const row = Math.floor(i / ahrPerRow);
const ax = 0.3 + col * 4.25;
const ay = 4.3 + row * 0.85;
s.addShape("roundRect", { x: ax, y: ay, w: 4.15, h: 0.75, rectRadius: 0.1, fill: { color: "0A2A0A" } });
s.addText("✦ " + item, { x: ax + 0.1, y: ay, w: 3.95, h: 0.75, fontSize: 8.5, color: "C8E6C9", valign: "middle" });
});
s.addText("Source: Robbins & Cotran (10e) | Fishman's Pulmonary Diseases", {
x: 0.3, y: 7.35, w: 12.7, h: 0.2, fontSize: 7.5, color: C.dim, align: "right"
});
}
// ═══════════════════════════════════════════════════════════════════════════
// Write file
// ═══════════════════════════════════════════════════════════════════════════
pres.writeFile({ fileName: "/home/daytona/workspace/asthma-ppt/Asthma_Pathophysiology_Flowchart.pptx" })
.then(() => console.log("Done: Asthma_Pathophysiology_Flowchart.pptx"))
.catch(e => { console.error(e); process.exit(1); });
You've run out of free credits. Sign up for a free account to continue using Orris.
This is a shared conversation. Sign in to Orris to start your own chat.