Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Finding Sources
Finding Sources
Finding Sources
I have more than enough material to write a comprehensive, well-sourced answer. Let me now compile the full explanation.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a syndrome of acute respiratory insufficiency caused by diffuse alveolar damage (DAD), triggered by a variety of direct (pulmonary) or indirect (extrapulmonary) insults. The pathophysiology unfolds across three overlapping phases.
Triggers and Initial Insult
Common precipitants fall into two categories:
| Direct (Pulmonary) | Indirect (Extrapulmonary) |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall trigger) |
| Aspiration of gastric contents | Severe trauma / polytrauma |
| Inhalation injury | Acute pancreatitis |
| Near-drowning | Massive transfusion (TRALI) |
| Diffuse pulmonary contusion | Burns, hemorrhagic shock |
- Robbins & Kumar Basic Pathology, p. 496
- Sabiston Textbook of Surgery, p. 940
Phase 1: Exudative Phase (Days 0-7) - The Core Mechanism
This is where the fundamental injury occurs. The sequence is:
1. Pattern Recognition and Macrophage Activation
The initial insult activates Toll-like receptors (TLRs) on alveolar type I (ATI) epithelial cells and resident alveolar macrophages. This triggers the release of pro-inflammatory cytokines, primarily TNF-alpha, IL-1beta, IL-6, IL-8, and IFN-beta.
2. Neutrophil Recruitment and Sequestration
- IL-8 and other PMN chemokines signal circulating neutrophils to marginate and extravasate from pulmonary capillaries into the alveolar interstitium and air spaces.
- Neutrophils are the central effector cells of alveolar-capillary membrane destruction.
- Once activated in the alveolar space, neutrophils release a destructive arsenal:
- Proteases (elastase, matrix metalloproteinases) - degrade the extracellular matrix and tight junctions
- Reactive oxygen species (ROS) - oxidant-mediated membrane injury
- Neutrophil extracellular traps (NETs) - promote coagulation and further cellular damage
- Platelet-activating factor (PAF) - amplifies vascular permeability
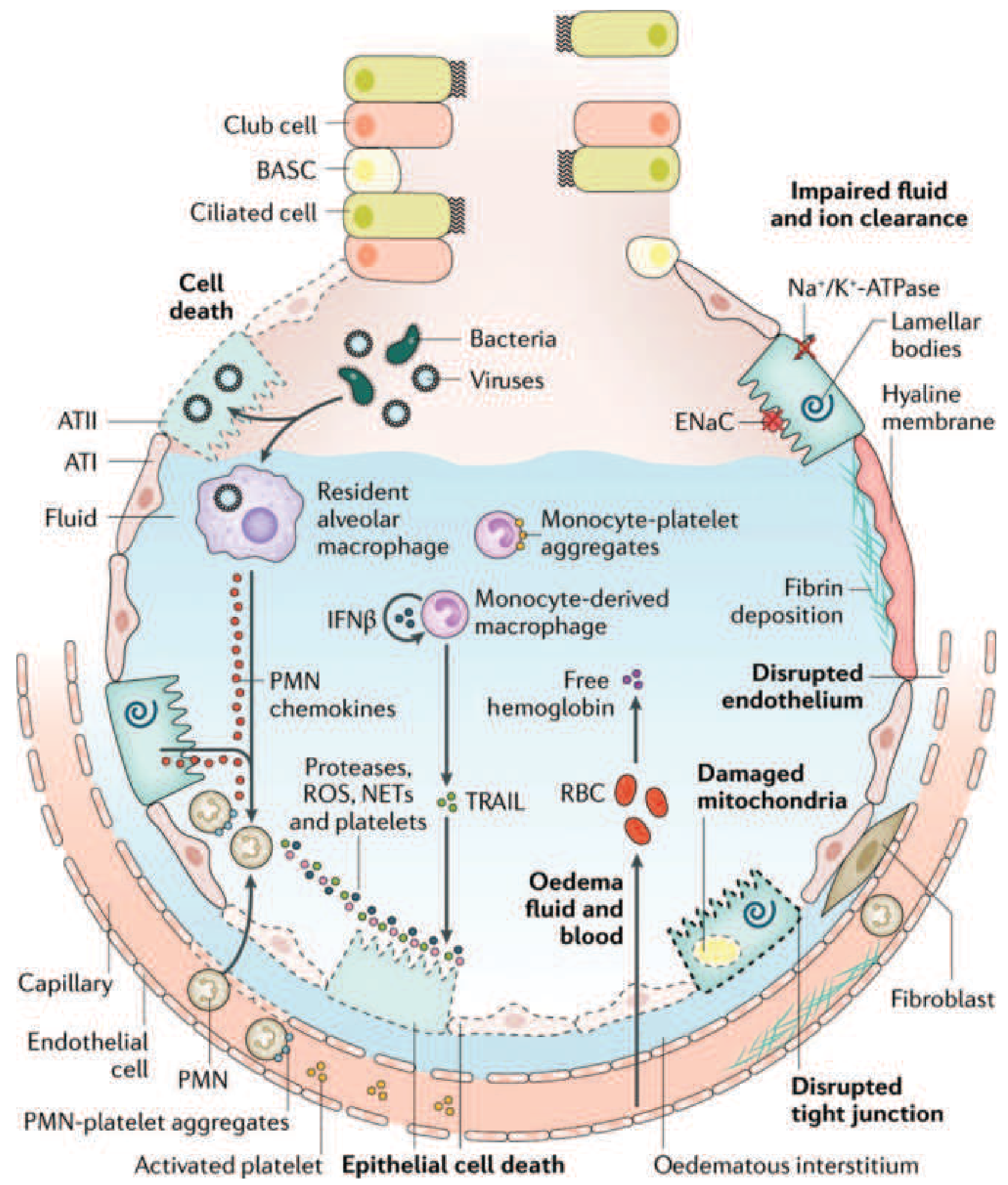
Figure: The injured alveolus in acute ARDS. PMNs (neutrophils) recruited from capillaries release proteases, ROS, and NETs, causing epithelial and endothelial cell death, edema fluid accumulation, fibrin deposition, and hyaline membrane formation. Na+/K+-ATPase and ENaC dysfunction impairs fluid clearance. - Harrison's Principles of Internal Medicine, 22E
3. Alveolar-Capillary Membrane Disruption
The combined neutrophil-mediated attack destroys:
- Type I alveolar epithelial cells (which cover 95% of the alveolar surface) - lost integrity leads to flooding
- Pulmonary capillary endothelium - tight junction disruption causes massive capillary leak
The result is the two defining pathological events:
- Non-cardiogenic pulmonary edema: Protein-rich fluid floods the alveolar space (hydrostatic pressure is normal; permeability is the problem)
- Impaired fluid and ion clearance: Loss of Na+/K+-ATPase and epithelial sodium channel (ENaC) function on type II pneumocytes impairs the active resorption of alveolar fluid
4. Surfactant Dysfunction
- Damaged type II pneumocytes cease producing surfactant
- Phospholipase A2 (released from neutrophils and, in pancreatitis-related ARDS, from the pancreas itself) enzymatically degrades existing surfactant
- Without surfactant, alveolar surface tension rises dramatically, causing alveolar collapse (atelectasis) and further reducing functional residual capacity (FRC)
5. Hyaline Membrane Formation and Fibrin Deposition
- Protein-rich exudate (containing fibrin, dead cells, and plasma proteins) lines the alveolar walls
- These coagulated deposits form hyaline membranes - the histologic hallmark of DAD
- Coagulation activation with fibrin deposition also occludes small vessels, contributing to dead-space ventilation and pulmonary hypertension
6. Ventilation-Perfusion (V/Q) Mismatch and Hypoxemia
The net result of these events:
- Consolidated, fluid-filled alveoli are perfused but not ventilated (intrapulmonary shunt)
- Decreased lung compliance (stiff lungs) increases the work of breathing
- Hypoxemia is refractory to supplemental oxygen because the shunt mechanism bypasses ventilated alveoli
- PaO2/FiO2 ratio falls below 300 (Berlin criteria: mild <300, moderate <200, severe <100)
Phase 2: Proliferative Phase (Days 7-21) - Repair Attempt
If the patient survives the exudative phase:
-
Neutrophil infiltrate transitions to a lymphocyte-predominant picture
-
Type II pneumocytes proliferate along alveolar basement membranes - they synthesize new surfactant and differentiate into type I cells
-
Alveolar exudates begin to organize and resorb
-
Many patients recover lung function during this phase, though dyspnea and hypoxemia may persist
-
Harrison's Principles of Internal Medicine 22E, p. 2344
Phase 3: Fibrotic Phase (>Day 21) - Maladaptive Repair
In a subset of patients, repair goes awry:
-
Alveolar exudates convert to alveolar-duct and interstitial fibrosis
-
Marked disruption of acinar architecture creates emphysema-like bullae
-
Intimal fibroproliferation in pulmonary microvessels causes progressive vascular occlusion and pulmonary hypertension
-
The physiologic consequences include decreased compliance, increased dead-space ventilation, increased pneumothorax risk, and long-term ventilator dependence
-
Harrison's Principles of Internal Medicine 22E, p. 2345
Timeline Summary

Figure: Time course of ARDS phases - Harrison's Principles of Internal Medicine, 22E
Additional Mechanistic Contributors
Coagulation dysregulation: ARDS activates both pro-coagulant and anti-fibrinolytic pathways within the lung. Fibrin thrombi in small vessels worsen V/Q mismatch and contribute to dead-space physiology.
Ventilator-induced lung injury (VILI): The heterogeneous distribution of ARDS (dependent consolidation, non-dependent sparing) means that tidal volume preferentially enters compliant regions. High tidal volumes cause:
-
Volutrauma (overdistention injury)
-
Barotrauma (pressure injury)
-
Atelectrauma (repetitive opening and collapse of unstable units)
-
Biotrauma (mechanical stress triggers cytokine release, worsening systemic inflammation and multi-organ dysfunction)
-
Goldman-Cecil Medicine, p. 1067
Key Mechanistic Summary
| Step | Event | Consequence |
|---|---|---|
| Insult (sepsis, trauma, aspiration) | TLR activation on ATI cells and alveolar macrophages | Pro-inflammatory cytokine storm |
| Cytokines (IL-8, TNF-alpha) | Neutrophil recruitment to alveoli | Effector cells concentrated at injury site |
| Neutrophil degranulation | Proteases, ROS, NETs | Alveolar-capillary membrane destruction |
| Barrier failure | Non-cardiogenic pulmonary edema | Alveolar flooding |
| Type II pneumocyte injury | Surfactant deficiency | Alveolar collapse, reduced FRC |
| Protein-rich exudate | Hyaline membrane formation | DAD histology, reduced diffusion |
| Shunt physiology + atelectasis | Refractory hypoxemia | PaO2/FiO2 <300 |
| Fibrin deposition, vascular occlusion | Pulmonary hypertension | Increased dead space |
| Maladaptive repair | Fibrosis (in some patients) | Irreversible lung damage |
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 312
- Robbins & Kumar Basic Pathology, Chapter 15
- Goldman-Cecil Medicine, Chapter 91
- Sabiston Textbook of Surgery, 11th Ed., Chapter 11
- Murray & Nadel's Textbook of Respiratory Medicine
This is a shared conversation. Sign in to Orris to start your own chat.