Organic acidurriosis biochemistry
Reading File
Finding Sources
Finding Sources
Searching PubMed
organic acidurias biochemistry metabolism
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
organic acidemia propionic methylmalonic metabolic pathway diagram
I now have comprehensive, authoritative content from multiple textbooks. Let me compose the complete answer:
Organic Acidurias — Biochemistry
Definition
Organic acidurias (organic acidemias) are a heterogeneous group of inborn errors of metabolism characterized by the accumulation of CoA-activated organic acid intermediates due to defects in the degradation of certain amino acids (and occasionally fatty acids or other substrates). They are distinguished from aminoacidopathies by the involvement of CoA-thioester intermediates rather than free amino acids.
The fundamental pathophysiological mechanism is accumulation of toxic substrates via primary blockage of a catabolic pathway — Goldman-Cecil Medicine describes this as the defining biochemical theme.
Biochemical Pathogenesis: Core Mechanisms
1. Substrate Accumulation → Toxicity
Defective enzymes cause upstream organic acid intermediates to accumulate. These CoA esters and their metabolites are directly toxic to multiple organ systems, particularly the mitochondria and CNS.
2. Inhibition of the Urea Cycle → Hyperammonemia
The accumulation of propionyl-CoA and related acyl-CoA esters inhibits N-acetylglutamate synthetase (NAGS), the first enzyme of the urea cycle. N-acetylglutamate is the allosteric activator of carbamoyl phosphate synthetase I (CPS-I). This intramitochondrial inhibition leads to secondary hyperammonemia — a hallmark of organic acidurias. — Bradley & Daroff's Neurology in Clinical Practice
3. Secondary Carnitine Deficiency
Organic acid metabolites are conjugated with carnitine (forming acylcarnitines) to facilitate their excretion. This depletes free carnitine stores, impairing long-chain fatty acid β-oxidation and compounding the energy deficit.
4. Mitochondrial Respiratory Chain Dysfunction
Accumulated toxic metabolites impair the electron transport chain, causing lactic acidosis, increased ROS, and decreased ATP production. This is particularly relevant in glutaric aciduria type I and MMA.
Major Organic Acidurias — Individual Biochemistry
A. Propionic Acidemia (PA)
| Feature | Detail |
|---|---|
| Defective enzyme | Propionyl-CoA carboxylase (biotin-dependent; subunits PCCA/PCCB) |
| Inheritance | Autosomal recessive |
| Substrate precursors | Isoleucine, valine, threonine, methionine, odd-chain fatty acids, uracil, thymine, cholesterol |
Biochemical pathway:
Propionyl-CoA →(propionyl-CoA carboxylase)→ Methylmalonyl-CoA →(methylmalonyl-CoA mutase)→ Succinyl-CoA → TCA cycle
In PA, the block is at step 1. Propionyl-CoA accumulates and is metabolized to 3-methylcitrate, propionic acid, and 3-hydroxypropionic acid — the urinary signature.
Key biochemical consequences:
- Severe metabolic ketoacidosis with high anion gap
- Hyperammonemia (via NAGS inhibition)
- Hyperglycinemia (propionyl-CoA inhibits the glycine cleavage system)
- Secondary carnitine deficiency (C3-carnitine elevated on newborn screen)
- Bone marrow suppression (neutropenia, thrombocytopenia)
- Cardiomyopathy (most common among OAs)
Diagnosis: ↑C3-acylcarnitine (propionylcarnitine); urine organic acids: ↑2-methylcitric acid, ↑3-hydroxypropionic acid
B. Methylmalonic Acidemia (MMA)
| Feature | Detail |
|---|---|
| Defective enzyme | Methylmalonyl-CoA mutase (adenosylcobalamin-dependent) — or defects in cobalamin metabolism |
| Inheritance | Autosomal recessive (X-linked in CblX form) |
| Same precursors as PA | Isoleucine, valine, threonine, methionine, odd-chain FAs |
Biochemical pathway:
L-Methylmalonyl-CoA →(methylmalonyl-CoA mutase + adenosylcobalamin)→ Succinyl-CoA
When mutase is deficient, methylmalonyl-CoA accumulates → methylmalonic acid excreted in urine and elevated in blood.
MMA shows extreme locus heterogeneity: pathogenic variants in:
- The mitochondrial enzyme MMUT (methylmalonyl-CoA mutase itself)
- Genes involved in cobalamin (B12) activation — absorption, transport, lysosomal release, or mitochondrial processing → defects in generating adenosylcobalamin (AdoCbl) and/or methylcobalamin (MeCbl)
The most common combined form is MMA + homocystinuria (CblC, MMACHC gene), where both MeCbl and AdoCbl are deficient, elevating both methylmalonic acid and homocysteine. — Goldman-Cecil Medicine
Severe B12 dietary deficiency can phenocopy the enzymatic defect.
Diagnosis: ↑C3-acylcarnitine; urine organic acids: markedly ↑methylmalonic acid; plasma: ↑methylmalonic acid, ±↑homocysteine
Complications: Progressive renal failure (distinctive to MMA), optic atrophy, "metabolic stroke" of basal ganglia
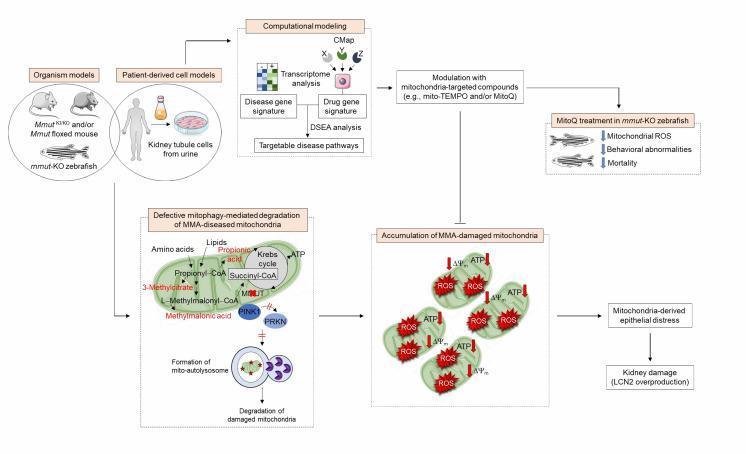
C. Isovaleric Acidemia (IVA)
| Feature | Detail |
|---|---|
| Defective enzyme | Isovaleryl-CoA dehydrogenase |
| Pathway | Leucine catabolism |
| Inheritance | Autosomal recessive |
Biochemical pathway:
Leucine → α-ketoisocaproate → Isovaleryl-CoA →(isovaleryl-CoA dehydrogenase)→ 3-Methylcrotonyl-CoA → … → Acetoacetate + Acetyl-CoA
Deficiency leads to accumulation of isovaleryl-CoA and its conjugation with glycine to form isovalerylglycine (the major urinary marker), and with carnitine → C5-acylcarnitine (newborn screening marker).
Clinical hallmark: "Sweaty feet" odor (isovaleric acid)
Biochemical markers: ↑C5-carnitine on MS/MS; urine: ↑isovalerylglycine, ↑3-hydroxyisovaleric acid
D. Glutaric Aciduria Type I (GA-1)
| Feature | Detail |
|---|---|
| Defective enzyme | Glutaryl-CoA dehydrogenase |
| Pathway | Lysine, hydroxylysine, and tryptophan catabolism |
| Inheritance | Autosomal recessive |
Biochemical pathway:
Lysine/Tryptophan → … → Glutaryl-CoA →(glutaryl-CoA dehydrogenase)→ Glutaconyl-CoA → Crotonyl-CoA → Acetyl-CoA
Deficiency → accumulation of glutaric acid and 3-hydroxyglutaric acid (the primary neurotoxin). These metabolites are particularly toxic to striatal neurons (putamen and caudate), causing acute encephalopathic crises triggered by febrile illness.
Diagnosis: Newborn screening: ↑glutarylcarnitine (C5DC); urine: ↑glutaric acid + ↑3-hydroxyglutaric acid
Neuroimaging: Frontotemporal hypoplasia ("open operculum"), bilateral striatal injury on MRI
E. Holocarboxylase Synthetase Deficiency (Multiple Carboxylase Deficiency)
| Feature | Detail |
|---|---|
| Defective enzyme | Holocarboxylase synthetase |
| Effect | Failure to biotinylate all four biotin-dependent carboxylases: propionyl-CoA carboxylase, pyruvate carboxylase, 3-methylcrotonyl-CoA carboxylase, acetyl-CoA carboxylase |
The biochemical consequence is a combined deficiency manifesting with accumulation of multiple organic acids: β-hydroxyisovaleric acid, β-methylcrotonylglycine, 3-hydroxypropionic acid, 3-methylcitrate.
Treatment: Pharmacological biotin supplementation (typically curative if given early)
F. Alkaptonuria
| Feature | Detail |
|---|---|
| Defective enzyme | Homogentisic acid oxidase |
| Pathway | Tyrosine degradation |
Phenylalanine → Tyrosine → 4-hydroxyphenylpyruvate → Homogentisic acid →(HGA oxidase)→ Maleylacetoacetate → Fumarate + Acetoacetate
Homogentisic acid (HA) accumulates, is excreted in urine (darkens on standing — oxidized to benzoquinone pigment), and is deposited in cartilage and collagenous tissue (ochronosis). — Lippincott Illustrated Reviews Biochemistry
Propionate Metabolism — Central Biochemical Hub
The propionate pathway is the convergence point for most major organic acidurias:
Isoleucine ─┐
Valine ─┤
Threonine ─┤──→ Propionyl-CoA ─→ [PA block here]
Methionine ─┤ ↓
Odd-chain FAs┘ Methylmalonyl-CoA ─→ [MMA block here]
↓ (adenosylcobalamin)
Succinyl-CoA → TCA cycle
Pathophysiology of Hyperammonemia in Organic Acidurias
This is a critical distinguishing mechanism from primary urea cycle disorders:
- Propionyl-CoA (and other acyl-CoA esters) inhibit N-acetylglutamate synthetase (NAGS)
- NAGS produces N-acetylglutamate, the obligate allosteric activator of CPS-I (carbamoyl phosphate synthetase I)
- Reduced N-acetylglutamate → reduced CPS-I activity → impaired urea cycle → hyperammonemia
- Unlike primary UCDs, plasma glutamine is not markedly elevated and orotic acid may be normal or mildly elevated (helps in differential diagnosis)
Newborn Screening — Biochemical Basis
Tandem mass spectrometry (MS/MS) detects acylcarnitine profiles in dried blood spots:
| Disorder | Acylcarnitine Marker |
|---|---|
| Propionic acidemia | C3 (propionylcarnitine) ↑ |
| Methylmalonic acidemia | C3 (propionylcarnitine) ↑ |
| Isovaleric acidemia | C5 (isovalerylcarnitine) ↑ |
| Glutaric aciduria type I | C5DC (glutarylcarnitine) ↑ |
| 3-Methylcrotonyl-CoA carboxylase def. | C5-OH ↑ |
C3 elevation distinguishes PA/MMA but not between them — second-tier urine organic acid analysis (↑2-methylcitric acid for PA; ↑methylmalonic acid for MMA) is used for confirmation.
Clinical Presentations by Pattern
| Pattern | Disorders |
|---|---|
| Neonatal intoxication (ketoacidosis, hyperammonemia, coma) | PA, MMA, IVA, MSUD |
| Macrocephaly + striatal injury on crisis | GA-1 |
| Chronic renal failure | MMA |
| Cardiomyopathy | PA (most common), MMA |
| "Sweaty feet" odor | IVA |
| Dark urine + arthritis | Alkaptonuria |
| Multiple organ involvement (responds to biotin) | Holocarboxylase synthetase deficiency |
Basal ganglia vulnerability is a shared feature — metabolic stroke-like episodes during febrile illness are characteristic, reflecting mitochondrial dysfunction in energy-dependent striatal neurons:
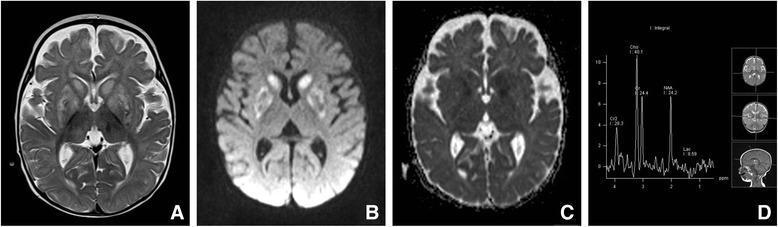
Treatment Principles
Treatment is disorder-specific but follows shared principles (Goldman-Cecil Medicine):
- Protein/precursor restriction — eliminate offending amino acids (e.g., Ile, Val, Thr, Met in PA/MMA; Leu in IVA; Lys/Trp in GA-1)
- Medical formula — provides non-restricted amino acids + calories
- Carnitine supplementation — promotes excretion of accumulated acyl-CoA metabolites
- Cofactor supplementation:
- Biotin (PA, holocarboxylase deficiency)
- Hydroxycobalamin (up to 30 mg/day SC) for cobalamin-responsive MMA; combined with betaine when homocysteine is elevated
- Ammonia management — N-acetylglutamate supplementation (carglumic acid) relieves CPS-I inhibition; nitrogen scavengers (sodium benzoate/phenylacetate) as acute therapy
- Organ transplantation — liver ± kidney transplant in severe MMA (reduces metabolic crises but does not fully prevent renal or neurological progression)
Sources: Goldman-Cecil Medicine, 26th ed. | Tietz Textbook of Laboratory Medicine, 7th ed. | Bradley & Daroff's Neurology in Clinical Practice | Lippincott Illustrated Reviews: Biochemistry, 8th ed. | Harrison's Principles of Internal Medicine, 22nd ed. | Emery's Elements of Medical Genetics and Genomics
This is a shared conversation. Sign in to Orris to start your own chat.