Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Here is a detailed explanation:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition and Overview
ARDS is defined by acute hypoxemia, bilateral opacities on chest imaging, and the absence of left atrial hypertension (i.e., it is non-cardiogenic pulmonary edema). Unlike congestive heart failure - where hydrostatic pressure forces fluid out of capillaries - in ARDS the edema fluid is exudative: the alveolar-capillary barrier loses its integrity, allowing protein-rich fluid to leak into the airspaces.
The Central Lesion: Alveolar-Capillary Barrier Breakdown
The entire mechanistic cascade converges on destruction of two cell layers:
- Pulmonary microvascular endothelium - loss of endothelial barrier integrity is both necessary and sufficient for ARDS to develop
- Alveolar epithelium (particularly Type I pneumocytes) - damage disrupts barrier integrity, prevents alveolar fluid clearance, and abolishes normal epithelial repair
Multiple mechanisms drive cell death in both layers: necrosis, apoptosis, activation of coagulation cascades, and mechanical stretch.
Step-by-Step Pathogenesis
1. Initiating Insult
ARDS is triggered by either a direct (pulmonary) or indirect (extrapulmonary) insult:
- Direct: pneumonia, aspiration, inhalational injury, pulmonary contusion
- Indirect: sepsis, pancreatitis, trauma, transfusion reactions (TRALI)
2. Innate Immune Activation and Cytokine Storm
Alveolar macrophages - the sentinels of the lung - respond to the initiating insult by releasing IL-1 and TNF-alpha. These cytokines:
- Activate pulmonary vascular endothelium
- Upregulate adhesion molecules (selectins, beta-2 integrins)
- Recruit and activate circulating neutrophils into the pulmonary microvasculature
3. Neutrophil Sequestration and Transmigration (Central Step)
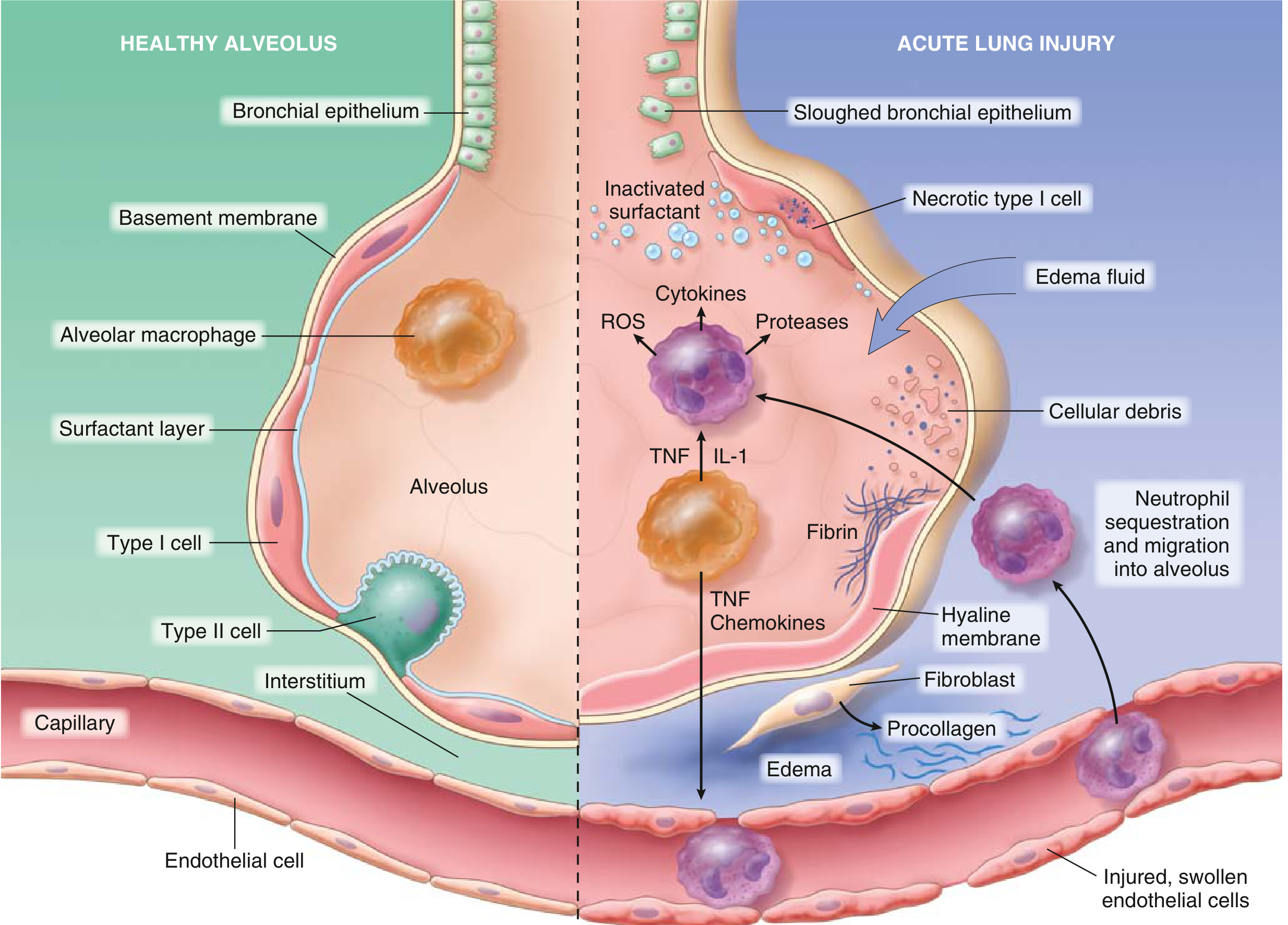
Neutrophils play the central role in ARDS pathogenesis. The mechanism of their accumulation is mechanically interesting:
- Pulmonary capillaries are narrower than neutrophil diameter, so neutrophils must deform to pass through
- Activated neutrophils become "stiff" (via actin cytoskeletal remodeling), cannot negotiate these narrow segments, and become sequestered in pulmonary capillaries
- One of the earliest signs is transient leukopenia as neutrophils are trapped in the lung before hypoxemia even develops
- Sequestered neutrophils then transmigrate across the alveolar-capillary membrane into the interstitium and alveolar space - and notably, this migration can occur without the usual requirement for L-selectin and beta-2 integrins
4. Neutrophil-Mediated Tissue Destruction
Once in the alveolar space, activated neutrophils unleash a battery of cytotoxic agents:
| Agent | Effect |
|---|---|
| Reactive oxygen species (ROS) | Oxidative damage to epithelial and endothelial membranes |
| Proteases (elastase, matrix metalloproteinases) | Degrade extracellular matrix, surfactant proteins, basement membranes |
| Defensins (cationic peptides) | Membrane disruption |
| Eicosanoids (leukotrienes, prostaglandins) | Amplify vascular permeability and inflammation |
| TNF-alpha, IL-1beta | Further amplify the inflammatory cascade |
| Neutrophil extracellular traps (NETs) | Trap pathogens but also damage host tissue |
The outcome of these assaults is determined by the balance between destructive factors and endogenous protective factors (antiproteases such as alpha-1 antitrypsin, antioxidants). When destruction outweighs protection, clinical ARDS results.
5. Surfactant Failure
- Leaked plasma proteins inactivate surfactant by competing with phospholipids at the air-liquid interface
- Neutrophil elastase degrades surfactant protein A
- The proportion of large (active) to small (inactive) surfactant aggregates falls due to decreased production and increased conversion
- Consequence: alveolar collapse (atelectasis), reduced lung compliance, and worsening hypoxemia
6. Coagulation Dysregulation
- The inflammatory milieu activates the coagulation cascade within the alveolar space
- Intravascular fibrin deposition and microvascular thrombosis further contribute to pulmonary hypertension and dead-space ventilation
- Fibrin in the alveolar lumen contributes to hyaline membrane formation
Pathophysiological Consequences
| Consequence | Mechanism |
|---|---|
| Profound hypoxemia | Right-to-left shunting through fluid-filled/collapsed alveoli |
| Reduced compliance ("stiff lungs") | Alveolar edema, surfactant failure, atelectasis |
| Pulmonary hypertension | Hypoxic vasoconstriction, intravascular fibrin, vessel compression by PEEP |
| Increased dead space | Microvascular thrombosis and obliteration |
| High work of breathing | Reduced compliance forces the respiratory muscles to generate greater effort |
Three Pathological Phases (Diffuse Alveolar Damage)
Phase 1 - Exudative (Days 1-7)
- Hyaline membranes (fibrin + cellular debris + surfactant remnants) line alveolar ducts
- Protein-rich edema floods the alveolar spaces
- Widespread epithelial disruption; necrosis of Type I pneumocytes
- Heavy neutrophil infiltration in interstitium and airspaces
- Macroscopic appearance: lungs are dark red, firm, airless, heavy
Phase 2 - Proliferative (Days 7-14)
- Type II pneumocytes proliferate vigorously to regenerate the alveolar lining
- Hyaline membranes are reorganized
- Neutrophils decrease; fibroblasts and procollagen appear
- Obliteration of pulmonary capillaries begins
Phase 3 - Fibrotic (>2 weeks)
- Fibrin-rich exudate undergoes organization
- Alveolar septal thickening and fibrosis
- Importantly, fibroproliferation may begin simultaneously with inflammatory injury (elevated N-terminal procollagen III peptide is detectable in BAL fluid as early as 24 hours after ARDS onset)
Ventilator-Induced Lung Injury (VILI) and Biotrauma
A critical concept: mechanical ventilation itself can worsen ARDS through two mechanisms:
- Volutrauma/barotrauma: High tidal volumes cause stress fractures in the alveolar-capillary membrane and basement membrane
- Biotrauma: High tidal volumes and zero PEEP trigger local cytokine release (TNF, IL-1, IL-6) that spills into the systemic circulation, potentially driving multiorgan failure - a phenomenon called biotrauma. Switching from a protective to an injurious ventilation strategy increases proinflammatory cytokines in the lungs and plasma within one hour.
This is why the ARDS Network lung-protective strategy (tidal volumes of 6 mL/kg ideal body weight) is a cornerstone of management.
Neutrophil-Platelet Interactions
An area of growing research: mutual activation of neutrophils and platelets amplifies lung injury in sepsis-associated ARDS. Platelet activation promotes neutrophil sequestration, and activated neutrophils in turn promote platelet aggregation, establishing a feedforward loop within the pulmonary microvasculature.
Summary Diagram of the Core Cascade
Initiating insult (sepsis, pneumonia, aspiration, trauma)
↓
Macrophage activation → IL-1, TNF-alpha
↓
Endothelial activation, neutrophil recruitment
↓
Neutrophil sequestration in pulmonary capillaries (stiffness → trapping)
↓
Transmigration into alveolar space
↓
ROS + proteases + cytokines released
↓
Alveolar epithelial + endothelial injury
↓
┌──────────────────────────────────────────────────────────────┐
│ Increased permeability → protein-rich edema │
│ Surfactant inactivation → atelectasis │
│ Coagulation activation → fibrin, hyaline membranes │
│ Type I pneumocyte death → impaired fluid clearance │
└──────────────────────────────────────────────────────────────┘
↓
Hypoxemia, reduced compliance, pulmonary hypertension (ARDS)
↓ (if ventilated injuriously)
Biotrauma → systemic cytokine dissemination → multiorgan failure
Sources: Robbins & Kumar Basic Pathology (p. 444); Murray & Nadel's Textbook of Respiratory Medicine (pp. 3146-3150); Fishman's Pulmonary Diseases and Disorders (pp. 2483-2484)
This is a shared conversation. Sign in to Orris to start your own chat.