***** Myocardial Infarction – (AP 16, API 17, TSI 19, TS 22, AP 24) a. Risk Factors & Etiopathogenesis b. Classification of Infarcts c. Morphology (Gross & Microscopy) d. Clinical Features & Complications e. Diagnosis of MI CASE – Dyspnoea & Chest Pain, Excessive Sweating, Pain radiating to Left Shoulder, Diabetic, Hypertensive, Troponins + in Blood Essay for 10 marks in pathology
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from Robbins Cotran (the authoritative pathology textbook). I have all the data I need to write a complete 10-mark essay. Let me compose the full answer:
Myocardial Infarction - Complete 10-Mark Essay (Pathology)
Case Analysis
The patient presents with dyspnoea, chest pain, diaphoresis (excessive sweating), pain radiating to the left shoulder, with background history of diabetes mellitus and hypertension, and positive serum troponins - this is a classic presentation of Acute Myocardial Infarction (AMI).
Definition
Myocardial infarction (MI), commonly called a "heart attack," is necrosis of cardiac muscle resulting from prolonged ischemia. The 2018 joint task force defines it as "acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
- Robbins & Kumar Basic Pathology, p. 334
- Robbins Cotran Kumar Pathologic Basis of Disease, p. 511
A. Risk Factors & Etiopathogenesis
Risk Factors
| Modifiable | Non-Modifiable |
|---|---|
| Hypertension | Age (risk rises progressively) |
| Diabetes mellitus | Male sex (females protected premenopausally) |
| Dyslipidemia (high LDL, low HDL) | Family history |
| Smoking / tobacco use | Genetic predisposition |
| Obesity | |
| Sedentary lifestyle | |
| Stress |
In the given case: The patient has diabetes + hypertension - both are established independent risk factors for atherosclerosis and MI. Studies show 90% of MI patients have at least one established risk factor prior to their first event.
Etiopathogenesis
The vast majority of MIs result from acute thrombosis within coronary arteries following disruption of an atherosclerotic plaque. The sequence is:
-
Plaque disruption - An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces, exposing subendothelial collagen and necrotic plaque contents to blood.
-
Platelet activation - Platelets adhere, aggregate, and are activated, releasing thromboxane A2, ADP, and serotonin, causing further platelet aggregation and vasospasm.
-
Coagulation activation - Tissue factor exposure activates the coagulation cascade, adding to the growing thrombus.
-
Occlusion - Within minutes, the enlarging thrombus may completely occlude the coronary artery lumen.
Angiography within 4 hours of MI onset demonstrates coronary thrombosis in almost 90% of cases.
Important: The culprit plaque often has less than 70% stenosis pre-event - it is vulnerable plaque with a large lipid core and thin fibrous cap that ruptures unpredictably.
In ~10% of cases, MI occurs without typical atherothrombosis - causes include:
- Coronary vasospasm (with or without atherosclerosis)
- Emboli from mural thrombi (e.g., atrial fibrillation) or valve vegetations
- Vasculitis, amyloid deposition, sickle cell disease
- Cocaine-induced vasospasm
B. Classification of Infarcts
1. By Depth (Extent)
| Type | Description | Cause |
|---|---|---|
| Transmural (Full-thickness) | Entire wall thickness involved; involves epicardium, myocardium, endocardium | Complete occlusion of a major coronary artery; associated with STEMI on ECG |
| Subendocardial (Non-transmural) | Limited to inner 1/3 to 1/2 of wall; inner myocardium is most vulnerable (furthest from epicardial supply) | Partial or transient occlusion; diffuse hypoperfusion; associated with NSTEMI |
2. By ECG Pattern (Clinically Used Classification)
| Type | ECG | Troponin | Description |
|---|---|---|---|
| STEMI (ST-Elevation MI) | ST elevation → Q waves | Positive | Transmural; complete occlusion |
| NSTEMI (Non-ST-Elevation MI) | ST depression / T-wave changes / normal | Positive | Subendocardial; incomplete occlusion |
| UA (Unstable Angina) | ST/T changes | Negative | No necrosis |
3. By Anatomical Location
| Coronary Artery | Territory Infarcted |
|---|---|
| Left Anterior Descending (LAD) - most common (~40-50%) | Anterior wall, anterior septum, apex |
| Right Coronary Artery (RCA) | Posterior/inferior wall, posterior septum, RV |
| Left Circumflex (LCX) | Lateral wall |
C. Morphology (Gross & Microscopy)
Gross Morphology - Time-Based Changes
| Time | Gross Appearance |
|---|---|
| 0 - 12 hours | No gross change visible; detected by TTC stain (pale unstained zone = infarct; brick-red = normal) |
| 12 - 24 hours | Reddish-blue discoloration from congestion and extravasated blood (dark mottling) |
| 1 - 3 days | Mottling with yellow-tan infarct center |
| 3 - 7 days | Hyperemic border; central yellow-tan softening; most vulnerable to rupture |
| 7 - 10 days | Maximally yellow-tan and soft; depressed red-tan margins |
| 10 - 14 days | Red-gray depressed infarct borders |
| 2 - 8 weeks | Gray-white scar, progressing from border toward core |
| > 2 months | Dense fibrous scar (complete) |
Triphenyl tetrazolium chloride (TTC) stain: Intact myocardium turns brick-red (preserved lactate dehydrogenase); infarcted tissue remains pale because dehydrogenases have leaked out through damaged membranes.
Microscopic Morphology - Time-Based Changes
| Time | Light Microscopy | Electron Microscopy |
|---|---|---|
| 0 - 0.5 hr | None | Myofibril relaxation; glycogen loss; mitochondrial swelling |
| 0.5 - 4 hrs | Usually none; wavy fibers at border (non-contractile dead fibers stretched by adjacent viable contracting myocardium) | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4 - 12 hrs | Onset of coagulative necrosis; edema; hemorrhage; nuclear pyknosis | - |
| 12 - 24 hrs | Ongoing coagulative necrosis; pyknosis; hypereosinophilic myocytes; marginal contraction band necrosis; early neutrophilic infiltrate | - |
| 1 - 3 days | Coagulative necrosis with loss of nuclei and striations; brisk neutrophilic infiltrate | - |
| 3 - 7 days | Disintegration of dead myofibers; dying neutrophils; early macrophage phagocytosis at border; early granulation tissue | - |
| 7 - 10 days | Well-developed phagocytosis; granulation tissue (most prominent) at margins | - |
| 10 - 14 days | Well-established granulation tissue with new blood vessels; collagen deposition begins | - |
| 2 - 8 weeks | Increased collagen deposition, decreased cellularity | - |
| > 2 months | Dense collagenous scar (complete) | - |
Key histological markers:
- Wavy fibers - earliest microscopic sign (0.5-4 hrs)
- Coagulative necrosis - hallmark (4-12 hrs)
- Contraction band necrosis - especially in reperfused infarcts (intense eosinophilic bands of hypercontracted sarcomeres due to calcium influx)
- Neutrophils - peak 1-3 days
- Macrophages - peak 5-10 days
- Granulation tissue - 7-14 days
- Dense scar - >2 months
Robbins Cotran Kumar, p. 513 (Table 12.5)
D. Clinical Features & Complications
Clinical Features
Symptoms:
- Chest pain - severe, crushing, pressure-like, substernal (most common); lasts >20-30 minutes (unlike angina)
- Radiation - to left arm/shoulder, jaw, neck, back, epigastrium (as in the case above)
- Diaphoresis (excessive sweating) - due to sympathetic activation
- Dyspnoea - due to LV dysfunction / pulmonary edema
- Nausea, vomiting - common
- Sense of impending doom
Silent MI: Up to 10-15% of MIs are clinically silent, especially in diabetics (impaired pain perception due to neuropathy) and elderly patients - relevant here as the patient has diabetes.
Signs:
- Tachycardia, may have bradycardia in inferior MI (vagal effect)
- S3 or S4 gallop
- Hypotension (cardiogenic shock in severe cases)
- Friction rub (pericarditis - 2-3 days after)
Complications (in order of timing)
| Complication | Timing | Details |
|---|---|---|
| Arrhythmias | Immediate (first hours/days) | Most common complication; leading cause of death pre-hospital; ventricular fibrillation, heart block, PVCs |
| Left ventricular failure / Pulmonary edema | Early | Due to loss of contractile myocardium |
| Cardiogenic shock | Early | Occurs when >40% LV mass is infarcted; high mortality |
| Myocardial rupture | 3-7 days (peak softening) | Three types: (1) Free wall rupture → hemopericardium → cardiac tamponade; (2) Interventricular septum rupture → acute VSD; (3) Papillary muscle rupture → acute MR |
| Pericarditis (Fibrinous) | 2-3 days (early) | Overlying epicarditis; friction rub; Dressler syndrome (autoimmune) at 2-10 weeks |
| Mural thrombus | Days-weeks | On akinetic wall → risk of systemic embolism (stroke) |
| Ventricular aneurysm | Weeks-months | Paradoxical bulge on systole; risk of mural thrombus, arrhythmias, heart failure |
| Progressive CHF / Ischemic cardiomyopathy | Long-term | Due to scar remodeling and progressive myocyte loss |
Robbins Cotran Kumar, p. 519
E. Diagnosis of MI
1. Cardiac Biomarkers (Most Important - as in the Case)
| Marker | Onset | Peak | Normalizes | Notes |
|---|---|---|---|---|
| Troponin I / Troponin T | 3-6 hrs | 24-48 hrs | 7-10 days (cTnI); 10-14 days (cTnT) | Gold standard - most sensitive and specific; present in the case |
| CK-MB (Creatine kinase-MB) | 3-6 hrs | 24 hrs | 48-72 hrs | Useful for reinfarction (re-elevation) |
| Myoglobin | 1-2 hrs | 4-8 hrs | 24 hrs | First to rise; non-specific |
| LDH (LDH1 > LDH2) | 24-48 hrs | 3-5 days | 7-14 days | "Flipped pattern" |
| SGOT (AST) | 6-12 hrs | 24-48 hrs | 3-5 days | Rarely used now |
In this case: Positive troponins confirm irreversible myocardial necrosis - diagnostic of MI.
2. ECG Changes
| Phase | Changes |
|---|---|
| Hyperacute (minutes-hours) | Tall, peaked T waves (first change) |
| Acute (hours) | ST-segment elevation (STEMI) or depression (NSTEMI); T-wave inversion |
| Evolving (days) | Q waves develop (pathological Q = >0.04 sec wide, >1/3 R height) |
| Old/Healed (weeks-months) | Persistent Q waves; T-wave normalization |
3. Imaging
- Echocardiography - Regional wall motion abnormalities; ejection fraction assessment
- Coronary angiography (Gold standard for anatomy) - identifies site of occlusion
- Cardiac MRI - late gadolinium enhancement shows scarring
- Radionuclide imaging (Thallium-201, Tc-99m sestamibi) - perfusion defects
4. Other Lab Findings
- Leukocytosis (neutrophilia) - reflects inflammatory response to necrosis
- Elevated ESR, CRP (acute phase response)
- Hyperglycemia (even in non-diabetics, due to stress response)
Case Summary (Applying Pathology to the Case)
| Feature | Pathological Basis |
|---|---|
| Chest pain radiating to left shoulder | Ischemic pain referred via T1-T4 dermatomes |
| Diaphoresis | Sympathetic nervous system activation |
| Dyspnoea | LV dysfunction → elevated LVEDP → pulmonary congestion |
| Diabetes | Accelerates atherosclerosis + increases risk of silent MI |
| Hypertension | Promotes atherosclerosis; increases cardiac afterload |
| Troponin positive | Sarcolemmal disruption → troponin release from necrotic myocytes |
Quick Summary Table for Exam
| Aspect | Key Point |
|---|---|
| Most common cause | Atherosclerotic plaque rupture + thrombosis |
| Most common vessel | LAD (anterior MI) |
| First gross change | Detectable by TTC stain at 2-3 hrs |
| Earliest microscopic sign | Wavy fibers (0.5-4 hrs) |
| Most vulnerable to rupture | 3-7 days (maximal softening) |
| Best diagnostic marker | Troponin I/T (gold standard) |
| Most common complication | Arrhythmias (leading cause of early death) |
| Most dangerous complication | Free wall rupture → cardiac tamponade (3-7 days) |
| Long-term complication | LV aneurysm, ischemic cardiomyopathy |
Sources: Robbins & Kumar Basic Pathology (p. 334-340) | Robbins, Cotran & Kumar Pathologic Basis of Disease (pp. 511-519)
Glioblastoma for 5 Mark
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I have everything needed for a comprehensive 5-mark essay on Glioblastoma. Here is the complete answer:
Glioblastoma - 5-Mark Essay (Pathology)
Definition & Introduction
Glioblastoma (GBM), IDH-wildtype, WHO Grade 4 is the most common and most malignant primary brain tumor in adults, arising from astrocytes. It accounts for approximately 50% of all primary malignant brain tumors (and >50% of all CNS malignancies). It is always considered a Grade 4 lesion with no lower-grade precursors, and carries a very poor prognosis.
- Robbins & Kumar Basic Pathology, p. 802
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1194
Epidemiology & Site
- Predominantly affects older adults in their 6th to 8th decades of life
- Location: Cerebral hemispheres - temporal, parietal, and frontal lobes; basal ganglia and thalamus
- De novo (primary) GBM is most common - arises without a preceding low-grade precursor
- Secondary GBM is rare - may arise from progression of lower-grade IDH-mutant astrocytoma
Pathogenesis & Molecular Genetics
GBM is defined by IDH-wildtype status (no IDH1/IDH2 mutation). It harbors multiple key genetic alterations:
| Genetic Alteration | Consequence |
|---|---|
| TERT promoter mutation | Telomerase activation → evasion of senescence |
| EGFR gene amplification | Activation of growth factor signaling |
| Combined +7 / -10 (gain chr.7, loss chr.10) | Most characteristic chromosomal change |
| Homozygous CDKN2A deletion | Loss of p16 → escape from growth controls |
| TP53 mutation | Resistance to apoptosis |
| MGMT promoter methylation | Silences DNA repair enzyme (predicts better response to chemotherapy with temozolomide) |
| PDGFR amplification | Additional growth factor pathway activation |
| VEGF overproduction | Drives microvascular proliferation (response to hypoxia) |
Even an adult diffuse astrocytoma without high-grade histology is still classified as GBM if it is IDH-wildtype and has TERT mutation, EGFR amplification, or +7/-10 copy number changes.
Morphology
Gross Appearance
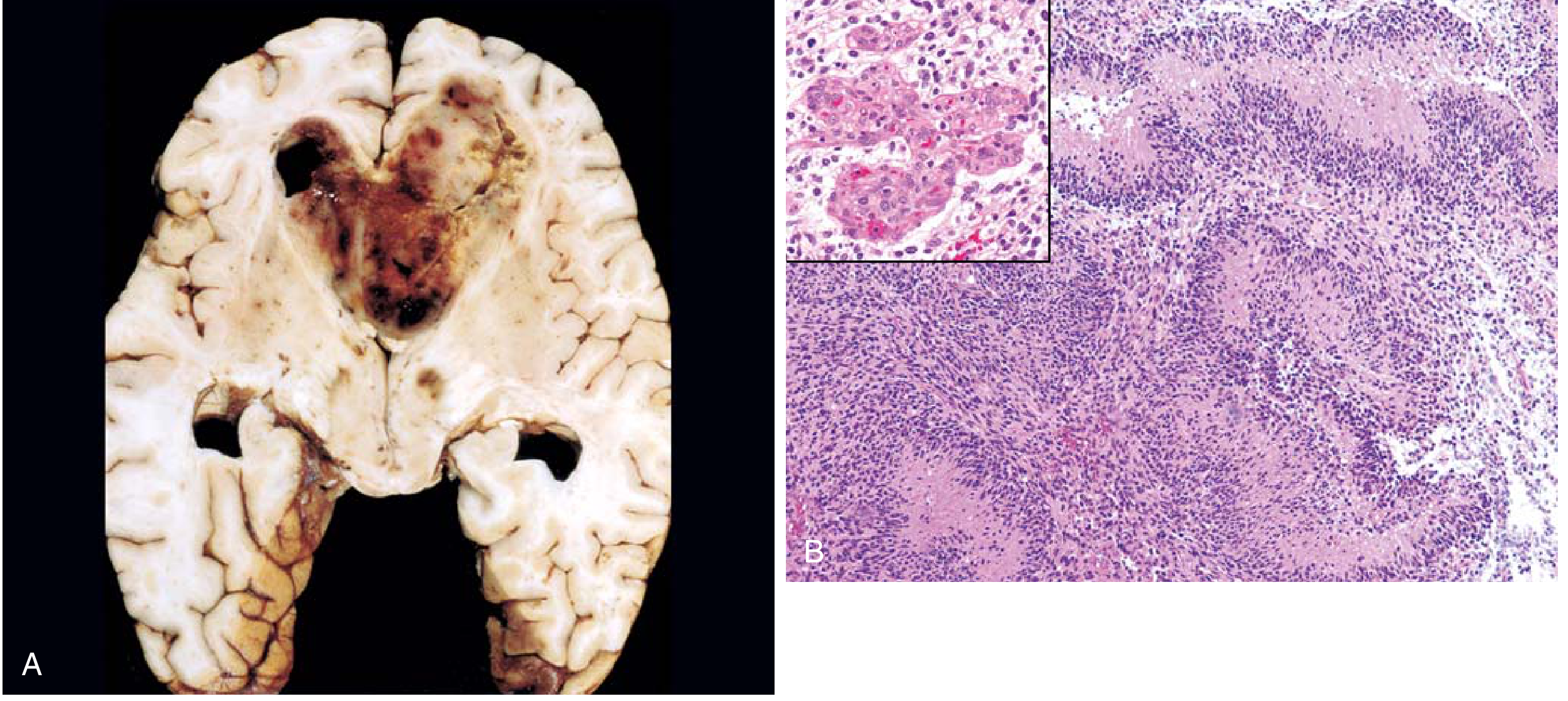
Fig. 21.31 - Glioblastoma. (A) Cross-section of brain showing necrotic, hemorrhagic, infiltrating mass. (B) Serpiginous palisading necrosis with microvascular proliferation (inset). - Robbins & Kumar
The gross appearance varies characteristically from region to region:
- Firm, white areas - solid tumor
- Soft, yellow areas - tissue necrosis
- Red/dark areas - hemorrhage
- Cystic areas - cystic degeneration
- Overall: necrotic, hemorrhagic, infiltrating mass
Butterfly glioma: Rapid infiltration through the corpus callosum leads to bilateral, symmetrical involvement of both hemispheres - a pathognomonic pattern.
Microscopic Appearance
The key diagnostic histological features are:
- High cellularity - densely packed, pleomorphic tumor cells
- Nuclear atypia - bizarre, pleomorphic nuclei
- Brisk mitotic activity - numerous mitoses
- Palisading (Pseudopalisading) necrosis - serpiginous/serpentine zones of necrosis surrounded by rows of tumor cells aligned perpendicular to the necrotic edge ("tumor nuclei palisade around necrotic zones") - pathognomonic
- Microvascular (endothelial) proliferation - tufts of multilayered cells bulging into vessel lumens ("glomeruloid bodies"), driven by VEGF released by hypoxic tumor cells - pathognomonic
- GFAP positive (glial fibrillary acidic protein) on immunohistochemistry
The two hallmark features that distinguish GBM from lower-grade gliomas are: Palisading necrosis + Microvascular proliferation.
Clinical Features
- Seizures - common presenting symptom
- Headache - severe, pulsating (raised ICP)
- Focal neurological deficits - depending on location (motor weakness, aphasia, visual field defects)
- Neurocognitive impairment - memory, personality changes
- Nausea, vomiting - raised ICP
- Rapid progression - symptoms develop over weeks
Imaging (MRI): Ring-enhancing lesion with central necrosis and surrounding edema (contrast enhancement due to leaky, abnormal blood-brain barrier). The ring-enhancing pattern on MRI is characteristic.
Diagnosis
- MRI with contrast - ring-enhancing lesion (imaging of choice)
- Biopsy + histopathology - gold standard for diagnosis
- Molecular profiling - IDH status (wildtype), TERT, EGFR, +7/-10, MGMT methylation
- GFAP immunostaining - confirms glial origin
Treatment & Prognosis
- Standard treatment (Stupp Protocol): Surgical resection + Radiotherapy + Temozolomide (alkylating agent)
- MGMT promoter methylation: Predicts better response to temozolomide chemotherapy
- Prognosis: Extremely poor - median survival is only 15-18 months even with aggressive treatment
Summary Table
| Feature | Glioblastoma (IDH-Wildtype, WHO Grade 4) |
|---|---|
| Cell of origin | Astrocyte |
| WHO grade | 4 (highest) |
| IDH status | Wildtype |
| Key molecular markers | TERT mut, EGFR amp, +7/-10, CDKN2A del |
| Age group | 6th-8th decade |
| Site | Cerebral hemispheres |
| Gross | Necrotic, hemorrhagic, infiltrating |
| Hallmark microscopy | Palisading necrosis + Microvascular proliferation |
| Imaging | Ring-enhancing lesion (MRI) |
| IHC | GFAP positive |
| Prognosis | Median survival 15-18 months |
Sources: Robbins & Kumar Basic Pathology (p. 802-803) | Robbins, Cotran & Kumar Pathologic Basis of Disease (pp. 1193-1196)
Pleomorphic adenoma
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Here is the complete, exam-ready answer on Pleomorphic Adenoma:
Pleomorphic Adenoma (Mixed Tumor)
Definition
Pleomorphic adenoma (also called "mixed tumor") is the most common benign tumor of the salivary glands, characterized by a mixture of ductal (epithelial), myoepithelial, and mesenchymal (stromal) elements. The term "pleomorphic" refers to this morphological heterogeneity (diverse tissue types), not to nuclear pleomorphism.
- Robbins & Kumar Basic Pathology, p. 543
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 697
Incidence & Site
| Feature | Detail |
|---|---|
| Most common salivary gland tumor | ~60% of all parotid tumors |
| Most common gland affected | Parotid gland (superficial lobe) - 80% |
| Second most common site | Submandibular gland |
| Minor salivary gland site | Hard palate (most common intraoral site) |
| Age group | 3rd-5th decade; any age |
| Sex | Slight female predominance |
"Rule of 70" (exam-relevant): 70% of salivary gland tumors are in the parotid; 70% of parotid tumors are pleomorphic adenoma; 70% are benign.
Pathogenesis / Molecular Genetics
- Cell of origin: All elements (epithelial, myoepithelial, mesenchymal-looking stroma) are believed to arise from myoepithelial or ductal reserve (stem) cells - hence the single-origin hypothesis despite the biphasic appearance.
- Radiation exposure increases risk.
- Key molecular alterations:
- PLAG1 gene overexpression (via chromosomal rearrangements, most commonly t(3;8)) - promotes growth factor receptor signaling
- HMGA2 gene mutation - in cases lacking PLAG1 overexpression
Morphology
Gross Appearance
- Rounded, well-demarcated, encapsulated mass (rarely exceeds 6 cm)
- Smooth surface (bosselated in some cases)
- Cut surface: gray-white with areas of:
- Myxoid (mucoid) - translucent gray-blue zones
- Chondroid (cartilage-like) - blue translucent areas
- Rarely: cystic degeneration or foci of ossification
- Capsule may be incomplete, especially on the palate, with tongue-like protrusions into surrounding tissue - important cause of recurrence after simple enucleation
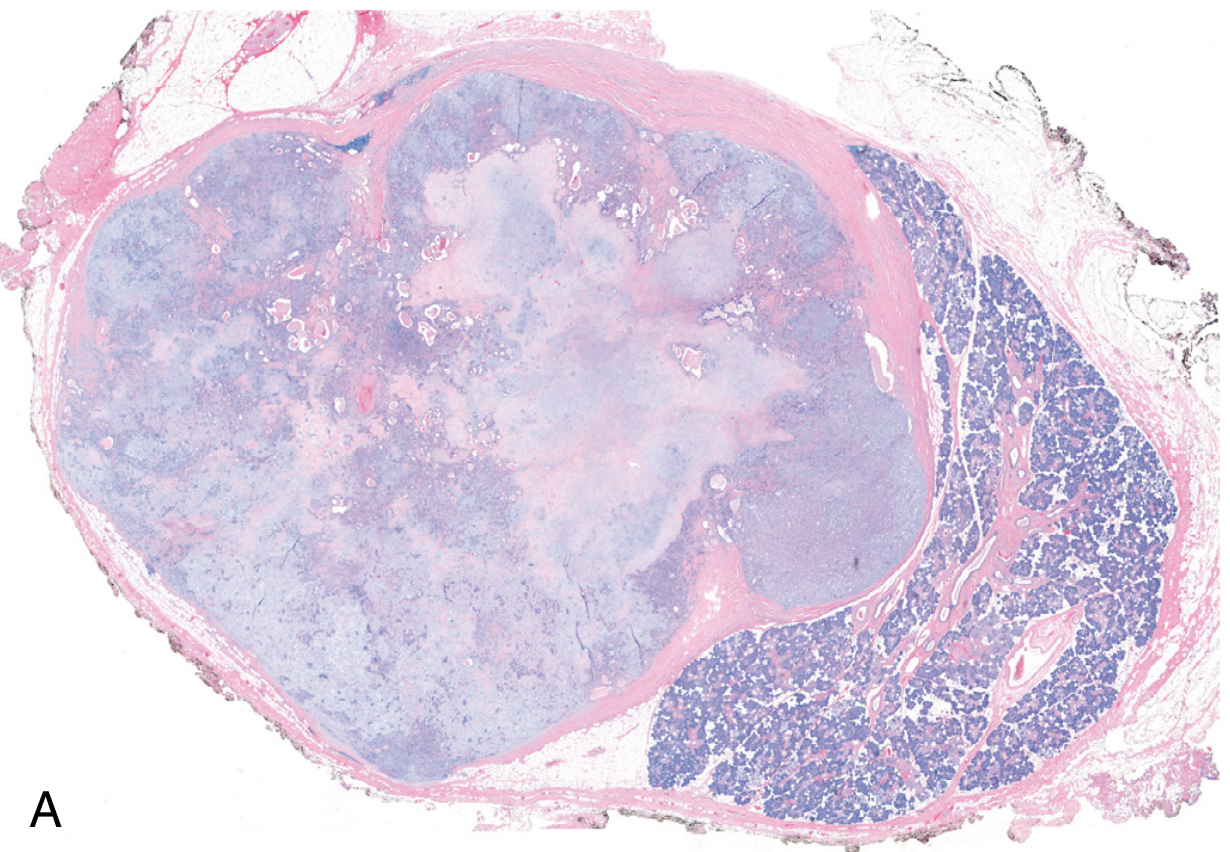
Fig. 16.20A - Low-power histologic view of pleomorphic adenoma: well-demarcated tumor (enclosed in capsule) with adjacent normal salivary gland parenchyma (right). Note the blue-purple chondromyxoid stroma within the tumor - Robbins Cotran Kumar
Microscopic Appearance
The hallmark is morphologic heterogeneity - a biphasic (dual-component) picture:
Component 1 - Epithelial/Myoepithelial elements:
- Cells arranged as ducts, acini, irregular tubules, strands, or sheets
- Ductal cells: cuboidal to columnar cells lining gland-like spaces
- Myoepithelial cells: small, deeply chromatic, spindle-shaped cells underlying ducts
- Islands of squamous epithelium may be present
- No significant nuclear atypia, no dysplasia, minimal mitoses
Component 2 - Stromal/Mesenchymal elements:
- Chondromyxoid matrix - loose myxoid tissue with islands of cartilage
- Rarely: foci of bone (osseous metaplasia)
- The stroma gives the "blue translucent" areas on gross
The ratio of epithelial to mesenchymal elements varies widely between tumors, but this does not affect prognosis.
Clinical Features
- Slow-growing, painless, mobile, discrete mass in the parotid region (below and in front of the ear / angle of jaw)
- Usually solitary; firm in consistency
- No facial nerve involvement (distinguishes from malignant parotid tumors)
- History of months to years of gradual enlargement
- No skin fixation or lymphadenopathy (benign)
Complications
1. Recurrence
- 25% recurrence after simple enucleation (due to incomplete capsule and tumor protrusions into surrounding tissue)
- Only 4% recurrence after wide local excision/superficial parotidectomy
- Multiple recurrences can lead to seeding of surgical field
2. Malignant Transformation - Carcinoma Ex Pleomorphic Adenoma
- Most serious complication
- Risk of malignant change increases with duration:
- < 5 years: ~2% risk
-
15 years: ~10% risk
- Malignant transformation usually takes the form of adenocarcinoma or undifferentiated carcinoma
- Highly aggressive - mortality 30-50% at 5 years
- Clinical clue: sudden rapid growth, pain, facial nerve palsy in a long-standing parotid mass
Diagnosis
| Investigation | Finding |
|---|---|
| FNAC (Fine Needle Aspiration Cytology) | Biphasic pattern - epithelial/myoepithelial cells in myxoid/chondromyxoid background; diagnostic |
| USG | Well-defined hypoechoic mass |
| MRI / CT | Well-encapsulated mass; MRI shows bright signal on T2 |
| Histopathology (post-excision) | Confirmatory - biphasic morphology as described above |
FNAC is the investigation of choice pre-operatively; incision biopsy is contraindicated (risk of seeding and facial nerve damage).
Treatment
- Superficial parotidectomy with preservation of the facial nerve - procedure of choice for parotid pleomorphic adenoma
- Simple enucleation is not recommended (high recurrence)
- For minor salivary gland tumors: wide local excision with clear margins
- No radiotherapy for benign disease (unless recurrent/unresectable)
Summary Table
| Feature | Pleomorphic Adenoma |
|---|---|
| Type | Benign |
| Most common salivary gland tumor | Yes (~60% of parotid tumors) |
| Cell of origin | Myoepithelial / ductal reserve cells |
| Key molecular change | PLAG1 overexpression; HMGA2 mutation |
| Gross | Encapsulated, bosselated; gray-white + myxoid/chondroid areas |
| Microscopy | Biphasic: epithelial/myoepithelial cells + chondromyxoid stroma |
| Clinical | Slow-growing, painless, mobile mass |
| Recurrence risk | 25% (enucleation); 4% (parotidectomy) |
| Malignant change | Carcinoma ex pleomorphic adenoma (2-10% risk) |
| Treatment | Superficial parotidectomy |
Sources: Robbins & Kumar Basic Pathology (p. 543) | Robbins, Cotran & Kumar Pathologic Basis of Disease (pp. 697-698) | Cummings Otolaryngology Head & Neck Surgery
Viral hepatitis
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive, authoritative content from both Robbins Cotran and Harrison's. Here is the complete essay:
Viral Hepatitis - Pathology Essay
Definition
Viral hepatitis is inflammation of the liver caused by a group of hepatotropic viruses - designated A, B, C, D, and E - each with distinct virology, transmission routes, clinical course, and outcomes. It ranges from acute self-limited illness to chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC).
The Five Hepatitis Viruses - Comparative Features
| Feature | Hep A (HAV) | Hep B (HBV) | Hep C (HCV) | Hep D (HDV) | Hep E (HEV) |
|---|---|---|---|---|---|
| Genome | ssRNA | Partially dsDNA | ssRNA | Circular defective ssRNA | ssRNA |
| Family | Picornaviridae | Hepadnaviridae | Flaviviridae | Deltaviridae | Hepeviridae |
| Transmission | Fecal-oral (food, water) | Parenteral, sexual, perinatal | Parenteral, IVDU | Parenteral (requires HBV) | Fecal-oral (water-borne) |
| Incubation Period | 2-6 weeks | 2-26 weeks | 4-26 weeks | Same as HBV | 4-5 weeks |
| Chronicity | Never | 5-10% | >80% | 10% co-infection; 90-100% superinfection | Immunocompromised only |
| Fulminant hepatitis | Rare (<0.1%) | Rare (0.1-1%) | Very rare | High risk with HBV | 20% mortality in pregnancy |
| HCC risk | No | Yes | Yes | Yes (with HBV) | No |
| Diagnosis | Serum IgM anti-HAV | HBsAg, anti-HBc antibodies; HBV DNA PCR | ELISA anti-HCV; HCV RNA PCR | IgM/IgG anti-HDV; HDV RNA | IgM anti-HEV; HEV RNA |
| Vaccine available | Yes | Yes | No | Prevented by HBV vaccine | Yes (some countries) |
Robbins, Cotran & Kumar - Table 18.3, p. 777
Pathogenesis
Liver injury in viral hepatitis is primarily immune-mediated, not directly cytopathic (except possibly HCV):
- HBV/HCV: CD8+ cytotoxic T cells recognize viral antigens on infected hepatocytes and destroy them. CD4+ helper T cells amplify the response. This cell-mediated immune attack on hepatocytes is the main mechanism of liver damage.
- HAV/HEV: Similar CD8+ T-cell cytolytic responses; viral shedding precedes liver injury.
- Extrahepatic manifestations (HBV): Immune complex (HBsAg-anti-HBs) deposition causes vasculitis (polyarteritis nodosa), glomerulonephritis, and cryoglobulinemia.
- HCV chronicity: Rapid mutation rate → viral quasispecies diversity → evades host immune containment. Also blocks type 1 interferon responses (NS3-4A protease).
Morphology
A. Acute Viral Hepatitis
Gross:
- Liver may be normal, enlarged (due to inflammation), or shrunken (in fulminant hepatic necrosis/"acute yellow atrophy")
Microscopy - General (all types):
| Feature | Description |
|---|---|
| Panlobular infiltration | Predominantly lymphocytes; plasma cells and eosinophils variably present |
| Hepatocyte injury | Necrosis and apoptosis; pigmented macrophages scavenging debris |
| Ballooning degeneration | Swollen, pale hepatocytes with cleared cytoplasm |
| Councilman (apoptotic) bodies | Acidophilic, shrunken, rounded hepatocytes = apoptotic hepatocytes (pyknotic nucleus + eosinophilic cytoplasm) - key feature |
| Kupffer cell hyperplasia | Reactive hyperplasia of sinusoidal macrophages |
| Mitotic figures / Rosette formation | Evidence of hepatocyte regeneration |
| Reticulin framework preserved | In uncomplicated acute hepatitis |
| Variable cholestasis | Bile plugs in canaliculi |
Patterns of necrosis in severe disease:
- Confluent necrosis - groups of hepatocytes
- Bridging necrosis - necrosis connecting portal tracts to central veins, or central vein to central vein (condensed reticulum + inflammatory debris = "bridge") - poor prognosis marker
- Panlobular/panacinar necrosis - entire lobule necrosed
- Massive hepatic necrosis (Fulminant hepatitis) - small, shrunken, soft liver; massive dropout of hepatocytes; extensive reticulin collapse
B. Chronic Viral Hepatitis
Defining feature: Portal lymphocytic (lymphoplasmacytic) inflammation with fibrosis
- Inflammatory cells cross the limiting plate → injury of periportal hepatocytes = "Interface hepatitis" (piecemeal necrosis)
- Fibrosis progression: portal/periportal → bridging fibrosis (portoportal) → cirrhosis
Virus-specific microscopic features:
| Virus | Distinctive Histological Feature |
|---|---|
| HBV | "Ground-glass" hepatocytes - ER swollen, filled with HBsAg; identified by Orcein or Aldehyde fuchsin stain; seen in chronic (not acute) HBV |
| HCV | Prominent lymphoid aggregates / lymphoid follicles in portal tracts; steatosis (especially genotype 3); focal bile duct injury |
| HDV | Microvesicular steatosis (occasional) |
| HEV | Marked cholestasis |
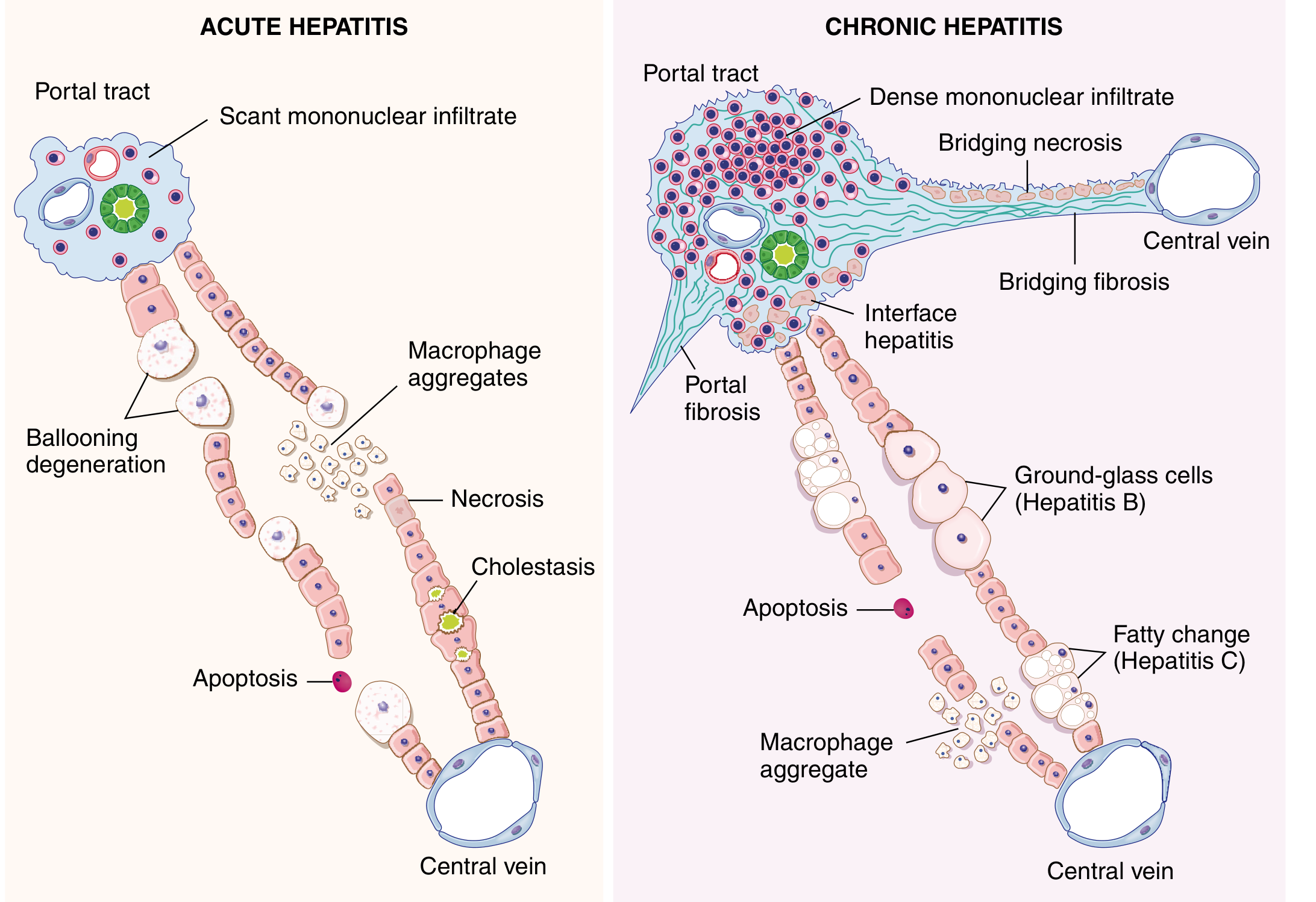
Fig. 18.14 - Diagrammatic representation of morphologic features of acute and chronic hepatitis - Robbins Cotran Kumar
Serological Markers of HBV (Most Tested in Exams)
| Marker | Significance |
|---|---|
| HBsAg | Hepatitis B surface antigen - first marker to appear; indicates infection (acute or chronic) |
| Anti-HBs | Antibody to HBsAg - indicates recovery and immunity; also appears after vaccination |
| HBcAg | Core antigen - found in liver (not routinely in serum) |
| Anti-HBc IgM | Acute HBV infection (appears early, short-lived) |
| Anti-HBc IgG | Past or chronic infection |
| HBeAg | Viral replication marker - indicates high infectivity |
| Anti-HBe | Seroconversion from HBeAg - indicates decreasing infectivity |
| HBV DNA | Most sensitive marker for active viral replication |
| "Window period" | HBsAg gone, Anti-HBs not yet appeared - only Anti-HBc IgM is positive |
Clinicopathologic Syndromes
1. Acute Symptomatic Hepatitis (4 Phases)
| Phase | Features |
|---|---|
| Incubation period | Virus-dependent (see table above); peak infectivity at end of this phase |
| Pre-icteric (prodromal) | Fever, malaise, anorexia, nausea, vomiting, RUQ pain, arthralgias (serum-sickness in HBV) |
| Icteric phase | Jaundice, dark urine, pale stools, hepatomegaly, ↑↑ ALT/AST |
| Convalescence | Gradual resolution of symptoms over weeks |
2. Acute Asymptomatic Infection with Recovery
- Subclinical; diagnosed by elevated transaminases or presence of antibodies
- Common with HAV and HBV
3. Chronic Hepatitis
- Defined as hepatitis persisting >6 months
- HBV: 5-10% of adults; HCV: >80% of all cases
- Progresses: chronic hepatitis → cirrhosis → HCC
4. Acute Liver Failure (Fulminant Hepatitis)
- Massive hepatic necrosis ("acute yellow atrophy")
- Encephalopathy, coagulopathy, jaundice
- HBV and HDV superinfection most common viral causes
- HEV: 20% mortality in pregnant women
- Pathology: small, shrunken liver; massive dropout of hepatocytes
5. Carrier State
- Persistent HBsAg positivity >6 months without active disease
- High-risk source of transmission
Clinical Features of Acute Hepatitis
- Constitutional: Fever, malaise, fatigue, anorexia, weight loss
- GI: Nausea, vomiting, abdominal pain (RUQ), diarrhea
- Jaundice: Icteric sclera, yellow skin
- Dark urine (bilirubinuria) and pale stools (reduced urobilinogen)
- Hepatomegaly - tender
- Raised ALT > AST (hepatocellular pattern)
Complications
| Complication | Virus |
|---|---|
| Chronic hepatitis | HBV, HCV, HDV |
| Cirrhosis | HBV, HCV (most common) |
| Hepatocellular carcinoma (HCC) | HBV (via HBx protein; integration), HCV (via chronic inflammation) |
| Fulminant hepatic failure | HBV, HBV+HDV, HEV in pregnancy |
| Extrahepatic: polyarteritis nodosa, GN | HBV |
| Extrahepatic: cryoglobulinemia, vasculitis | HCV |
| Aplastic anemia | HAV (rare) |
Summary - "ABCDE" Mnemonic
| Hepatitis | Key Fact |
|---|---|
| A | Acute only; fecal-oral; never chronic; vaccine available |
| B | Blood-borne; ground-glass cells; 5-10% chronic; HCC risk; vaccine available |
| C | Most common cause of chronic hepatitis (>80%); lymphoid aggregates + steatosis; no vaccine |
| D | Delta virus - defective; needs HBV; worst outcome in superinfection |
| E | Enteric (fecal-oral); deadly in pregnancy (20%); no chronicity in immunocompetent |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (pp. 777-779, Table 18.3) | Harrison's Principles of Internal Medicine 22nd Edition (pp. 2691-2694)
This is a shared conversation. Sign in to Orris to start your own chat.