Pharmacology of testosterone
Reading File
Finding Sources
Finding Sources
Searching PubMed
testosterone pharmacology mechanism action
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive data from multiple authoritative textbooks. Let me compile the full answer.
Pharmacology of Testosterone
1. Synthesis and Secretion
Testosterone is the most important androgen secreted by the testis. In men, approximately 8 mg is produced daily - about 95% by the Leydig cells (interstitial cells) under LH stimulation, and only 5% by the adrenals. The pathways of synthesis are shared with the adrenal gland and ovary, proceeding through cholesterol -> pregnenolone -> progesterone -> androstenedione -> testosterone via a series of P450 enzymes.
Women also have testosterone in plasma (approximately 0.03 mcg/dL), derived roughly equally from the ovaries, adrenals, and peripheral conversion. Normal male plasma levels are about 0.6 mcg/dL, declining after age 50.
The hypothalamic-pituitary-gonadal axis regulates testosterone:
- GnRH (pulsatile) -> LH and FSH from the pituitary
- LH stimulates Leydig cell testosterone synthesis
- Testosterone (and DHT and inhibin from Sertoli cells) exert negative feedback on pituitary FSH secretion
2. Transport and Protein Binding
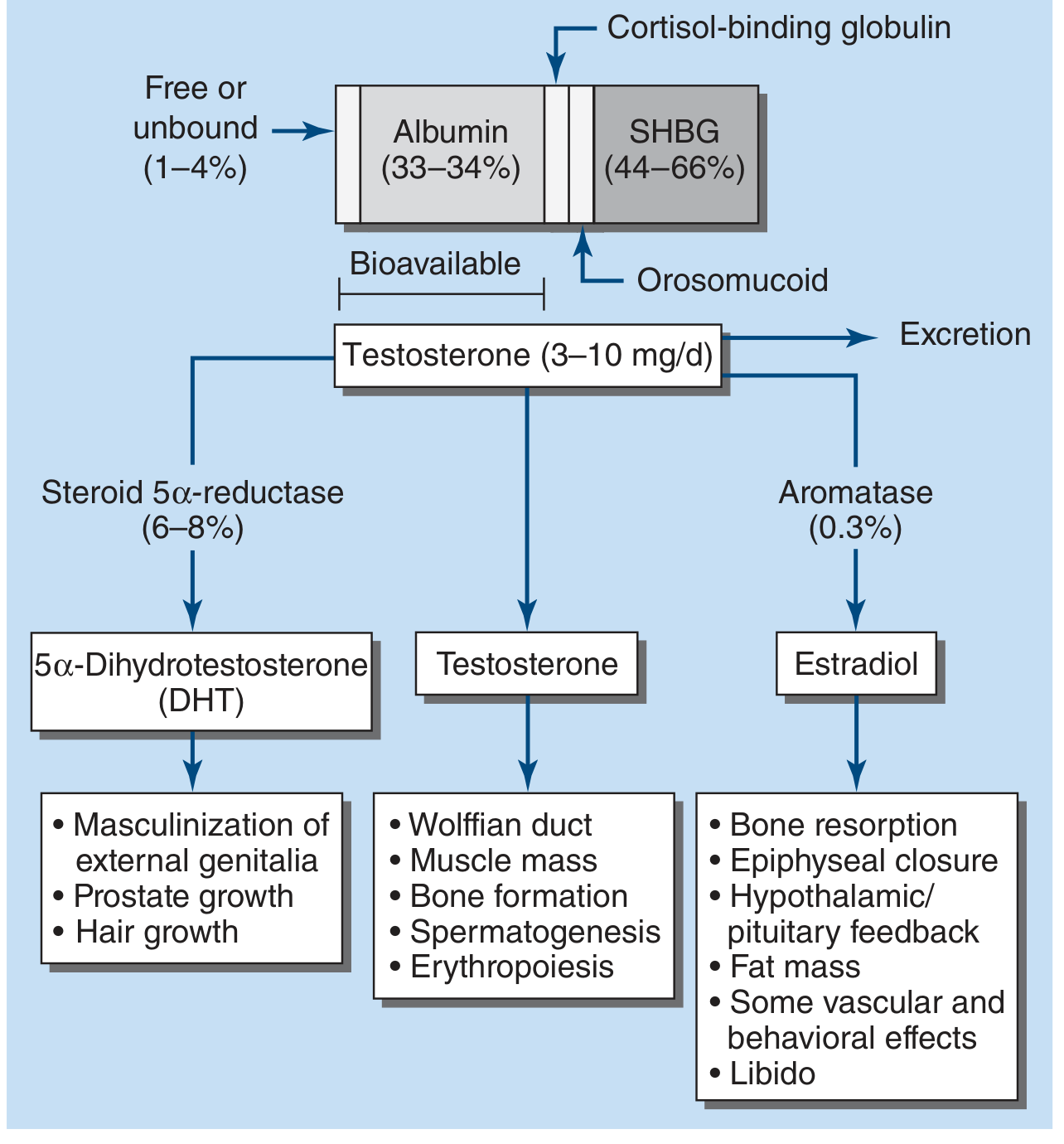
In plasma, testosterone is distributed as:
- ~65% bound to sex hormone-binding globulin (SHBG) - high-affinity binding
- ~33% bound to albumin (lower affinity, multiple allosteric binding sites)
- ~2% free (unbound) - biologically active fraction
- Small amount bound to cortisol-binding globulin (orosomucoid)
The "bioavailable" fraction includes the free testosterone plus albumin-bound testosterone.
Factors affecting SHBG:
- Increased by: estrogens, thyroid hormone, liver cirrhosis, HIV/hepatitis, aging, certain anticonvulsants
- Decreased by: androgens, growth hormone, obesity, diabetes, hypothyroidism, nephrotic syndrome
3. Mechanism of Action
Testosterone acts through both genomic and non-genomic pathways, and also through its active metabolites:
3a. Active Metabolites (the "prohormone" concept)
Testosterone is a prohormone converted in target tissues to two principal active forms:
| Metabolite | Enzyme | Key Tissues | Functions |
|---|---|---|---|
| 5α-Dihydrotestosterone (DHT) | 5α-reductase | Prostate, skin, genital skin | Masculinization of external genitalia, prostate growth, hair follicle effects |
| 17β-Estradiol | CYP19 (aromatase) | Adipose, liver, hypothalamus, bone | Bone resorption, epiphyseal closure, hypothalamic feedback, fat mass, libido, vascular and behavioral effects |
DHT has higher affinity for the androgen receptor (AR) than testosterone itself. Actions on Wolffian structures, skeletal muscle, erythropoiesis, and bone do not require conversion to DHT. The conversion of testosterone to DHT is necessary for masculinization of the urogenital sinus and genital tubercle, but not obligatory for muscle, bone, or hematopoiesis effects.
DHT can be further converted to:
- 5α-androstane-3β,17β-diol - a high-affinity agonist of estrogen receptor β
- 5α-androstane-3α,17β-diol - a modulator of GABA receptors
3b. Genomic (Nuclear Receptor) Pathway
- Testosterone (or DHT) diffuses into the target cell and binds the cytoplasmic androgen receptor (AR)
- The ligand-receptor complex undergoes conformational change, releases heat shock proteins, and translocates to the nucleus
- The AR complex dimerizes and binds androgen response elements (AREs) on DNA
- This augments gene transcription and ultimately new protein synthesis (anabolic, virilizing effects)
3c. Non-genomic Effects
Some effects of androgens may be non-genomic, acting through rapid signaling pathways at the cell membrane or through GABA receptor modulation. The 5α-androstanediol metabolite modulates GABA-A receptors, contributing to behavioral/neurological effects.
4. Physiologic Effects
| System | Effects |
|---|---|
| Sexual development | Penile and scrotal growth, secondary sex characteristics, deepening of voice, pubic/axillary/beard hair |
| Musculoskeletal | Skeletal growth, epiphyseal closure (acceleration), increased lean body mass, increased muscle mass and strength |
| Bone | Increased bone mineral density; estradiol (via aromatization) mediates epiphyseal closure and inhibits bone resorption |
| Erythropoiesis | Stimulates renal erythropoietin secretion -> increased RBC production and hematocrit |
| Skin/sebaceous | Thicker, oilier skin; increased sebum secretion (acne risk) |
| Liver (metabolic) | Decreased HDL, increased LDL; reduced SHBG; increased clotting factors, triglyceride lipase, α1-antitrypsin, haptoglobin |
| Reproductive | Prostate and seminal vesicle growth; local high concentrations required for spermatogenesis |
| CNS/behavior | Libido, mood, energy, spatial cognition; (via estradiol) hypothalamic-pituitary feedback |
5. Metabolism and Excretion
Testosterone is metabolized predominantly in the liver via 5α and 5β-reductases, 3α- and 3β-hydroxysteroid dehydrogenases, and 17β-HSD to inactive metabolites (androsterone, etiocholanolone, DHT, 3α-androstanediol). These undergo glucuronidation or sulfation and are excreted renally.
Some peripheral metabolism also occurs in the prostate and skin. The characteristic Δ4-ketone A ring of testosterone is reduced in the liver, leading to inactive androsterone and etiocholanolone conjugates.
6. Pharmaceutical Formulations
Oral crystalline testosterone is rapidly absorbed but undergoes extensive first-pass hepatic metabolism (only ~1/6 of the dose remains active). Various formulations have been developed to overcome this:
| Formulation | Example | Route | Key Features |
|---|---|---|---|
| 17α-Alkylated derivatives | Methyltestosterone, fluoxymesterone, oxandrolone | Oral | Resistant to first-pass; hepatotoxic - cholestatic jaundice, peliosis, adenoma risk. Only indicated for hereditary angioedema (C1 esterase deficiency). |
| Esterified (injectable) | Testosterone enanthate, cypionate (200 mg IM) | IM (oil depot) | Hydrolyzed at injection site; enanthate/cypionate last ~2 weeks (peaks day 1, troughs by day 14); biweekly causes mood/libido fluctuations - weekly dosing reduces this. |
| Long-acting injectable | Testosterone undecanoate (750 mg IM) | IM | Priming at 0 and 4 weeks, then every 10-14 weeks; stable levels; watch for POME (pulmonary oil microembolism) - observe for 30 min post-injection. |
| Transdermal gels/patches | Testosterone gel 1% (5-10 g/d) | Topical | Convenient; high DHT:testosterone ratio; risk of transfer to partners/children via skin contact - wash site before contact. |
| Oral testosterone undecanoate | Jatenzo (newer formulation) | Oral (with fat) | Absorbed via lymphatics, bypassing first-pass. Check levels 6-8 h post-dose. Higher DHT:T ratios. |
| Subcutaneous pellets | Testopel | SubQ implant | Last 3-4 months; drawback is need for incision, extrusion risk, fibrosis. |
| Intranasal gel | Natesto | Intranasal (11 mg TID) | Avoid nasal irritation, epistaxis, rhinorrhea. |
Target serum testosterone for replacement: mid-normal range (~400-750 ng/dL)
7. Clinical Indications
- Male hypogonadism (primary or secondary) - the major indication; restores secondary sex characteristics, libido, bone density, muscle mass, erythropoiesis, mood
- Delayed puberty in boys - short-term, with careful monitoring of bone age
- Hereditary angioedema (C1 esterase deficiency) - oral 17α-alkylated androgens stimulate hepatic C1-inhibitor synthesis
- Gynecologic uses - danazol (weak androgen) for endometriosis; androgens occasionally combined with estrogens in postmenopausal HRT
- Cachexia/wasting - in HIV-infected men with weight loss and low testosterone (systematic reviews confirm improvement in lean body mass and strength)
- Anemia in aplastic anemia (though EPO/CSFs now preferred due to hepatocellular carcinoma risk)
- Gender-affirming hormone therapy in transgender men
8. Adverse Effects
In women: Hirsutism, acne, amenorrhea, clitoral enlargement, voice deepening (>200-300 mg/month threshold, though some women are more sensitive). Progestational activity of some androgens causes endometrial bleeding on discontinuation.
In men on replacement therapy: Acne, sleep apnea, erythrocytosis/polycythemia, gynecomastia, azoospermia (supraphysiologic doses), decreased testicular volume, sodium and water retention (edema risk in heart/kidney disease).
17α-Alkylated androgens specifically:
- Cholestatic jaundice (reversible on stopping)
- Peliosis hepatis
- Hepatocellular adenoma and carcinoma
- Decreased HDL, increased LDL
- Behavioral: psychological dependence, increased aggressiveness, psychosis
In children: Premature epiphyseal closure -> short stature; CNS effects on sexual development centers (especially in females).
In fetuses: Maternal androgen use -> masculinization of female fetus, undermasculinization of male fetus.
9. Contraindications
- Pregnant women or women who may become pregnant
- Carcinoma of the prostate or male breast
- Infants and young children (except in specific circumstances)
- Severe/uncontrolled heart failure, recent MI, stroke, or acute coronary syndrome (within 4 months)
- Caution with renal or cardiac disease prone to edema
10. Monitoring (TRT)
Per Harrison's (22E) guidelines:
- Testosterone levels at 3-6 months, then annually (target 400-750 ng/dL)
- Hematocrit at baseline, 3-6 months, then annually - hold if >54%
- PSA + digital rectal exam at 3-6 months and per prostate cancer screening guidelines (men ≥55 years)
- Refer urology if PSA rises >1.4 ng/mL in first 12 months or PSA >4 ng/mL
- Bone mineral density after 1-2 years in hypogonadal men with osteoporosis or low-trauma fracture
11. Antiandrogens and Androgen Suppression
Several classes of drugs antagonize testosterone:
| Drug Class | Examples | Mechanism |
|---|---|---|
| GnRH agonists (continuous) | Leuprolide, goserelin, nafarelin | Continuous (non-pulsatile) administration desensitizes GnRH receptors -> LH/FSH suppression -> castrate testosterone levels |
| GnRH antagonists | Degarelix, relugolix | Competitive inhibition of GnRH receptors; faster onset than agonists |
| 5α-reductase inhibitors | Finasteride, dutasteride | Block conversion of testosterone to DHT; used in BPH, androgenetic alopecia, prostate cancer chemoprevention |
| Androgen receptor antagonists | Flutamide, bicalutamide, enzalutamide | Compete for binding to cytosolic AR; block nuclear translocation and gene activation |
| Steroidal antiandrogens | Spironolactone, cyproterone acetate | Competitive AR blockade; spironolactone also inhibits androgen synthesis |
12. Selective Androgen Receptor Modulators (SARMs)
SARMs are nonsteroidal AR ligands with tissue-selective actions. They bind the AR and induce specific conformational changes that modulate protein-protein interactions between AR and its coregulators, achieving tissue-specific gene expression. The goal is agonist activity on muscle and bone (anabolic) with antagonist or sparing effects on the prostate. Nonsteroidal SARMs are not substrates for 5α-reductase or aromatase. Several have advanced to phase 3 trials for muscle wasting, osteoporosis, and androgen deprivation in prostate cancer.
Sources: Katzung's Basic and Clinical Pharmacology (16e), Harrison's Principles of Internal Medicine (22e, 2025), Goodman & Gilman's Pharmacological Basis of Therapeutics, Costanzo Physiology (7e), Medical Physiology.
This is a shared conversation. Sign in to Orris to start your own chat.