Maple syrup urine disease
Reading File
Finding Sources
Searching PubMed
"maple syrup urine disease"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
I have enough comprehensive information to write a thorough response. Let me now compile the full answer.
Maple Syrup Urine Disease (MSUD)
Overview
Maple Syrup Urine Disease is a rare autosomal recessive inborn error of branched-chain amino acid (BCAA) metabolism with an incidence of approximately 1:185,000-200,000 live births. The name comes from the characteristic sweet, caramel-like odor of the urine (and sometimes cerumen) caused by accumulating branched-chain keto acids.
Pathophysiology
BCAAs - leucine, isoleucine, and valine - are essential amino acids. After transamination to their corresponding alpha-keto acids, they are normally decarboxylated by the mitochondrial branched-chain alpha-keto acid dehydrogenase (BCKD) complex.
This complex has four subunits:
- E1α and E1β - thiamine pyrophosphate (TPP)-dependent decarboxylase
- E2 - dihydrolipoyl transacylase
- E3 - dihydrolipoyl dehydrogenase (shared with pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase complexes)
A defect in any of these components causes MSUD. The result is toxic accumulation of BCAAs and their keto acids in blood and urine.
Leucine is the primary neurotoxic culprit - it causes cerebral edema and is responsible for the progressive neurologic deterioration. Isoleucine is responsible for the characteristic maple syrup odor (via its keto acid, 2-keto-3-methylvaleric acid, which undergoes racemization to produce L-alloisoleucine - the pathognomonic marker).
- Lippincott Biochemistry 8th ed., p. 768
- Tietz Textbook of Laboratory Medicine 7th ed.
Clinical Forms
| Type | BCKD Activity | Features |
|---|---|---|
| Classic (neonatal) | Near zero | Most severe; symptoms within days of birth |
| Intermediate | Up to 30% normal | Milder symptoms; onset infancy to adolescence |
| Intermittent | Variable | Episodes triggered by high protein intake or catabolism; may be normal between episodes |
| Thiamine-responsive | Partial | Responds to pharmacologic doses of thiamine (vitamin B1) |
| E3 deficiency | E3 absent | Combined deficiency of BCKD, pyruvate dehydrogenase, and alpha-ketoglutarate dehydrogenase; lactic acidosis is also present |
- Tietz Textbook of Laboratory Medicine 7th ed.
Clinical Presentation
Classic neonatal MSUD:
- Normal birth and uneventful first 24-48 hours (as BCAAs build up from dietary protein)
- Feeding difficulties and vomiting - first week of life
- Lethargy, hypotonia/hypertonia progressing to decerebrate rigidity
- Seizures
- Respiratory irregularities
- Coma if untreated
- Maple syrup/caramelized sugar odor of urine (and ear wax)
- Ketonuria (positive urine ketones)
- Hypoglycemia
- Fatal within weeks if untreated; survivors have severe intellectual disability
Milder/intermittent forms:
-
Recurrent vomiting, ataxia, lethargy
-
Developmental delay, seizures
-
Decompensation triggered by illness, fasting, or high protein intake
-
May return to near-normal between episodes but accumulate neurologic deficits
-
Henry's Clinical Diagnosis and Management by Laboratory Methods
-
Kaplan and Sadock's Synopsis of Psychiatry
Diagnosis
Screening:
- Newborn screening (tandem mass spectrometry): elevated leucine and isoleucine; normal phenylalanine distinguishes MSUD from PKU
- Patients with milder forms can be missed on screening
Urine tests:
- 2,4-Dinitrophenylhydrazine (DNPH) test - forms a yellow/white precipitate with alpha-keto acids (positive in MSUD, also positive in PKU, histidinemia, and other ketoacidurias; must exclude simple ketonuria first)
- Urine organic acid analysis shows characteristic branched-chain keto acids and 2-hydroxyisovaleric acid during decompensation
Confirmatory:
- Plasma amino acid analysis - markedly elevated leucine (usually most prominent), isoleucine, and valine, PLUS the pathognomonic L-alloisoleucine (a stereoisomer not normally present)
- Gas chromatography / thin-layer chromatography / NMR spectroscopy of urine
- BCKD enzyme activity in leukocytes or cultured skin fibroblasts (classic MSUD shows little or no activity)
Prenatal: Genetic testing available; most affected individuals are compound heterozygotes
- Tietz Textbook of Laboratory Medicine 7th ed.
- Henry's Clinical Diagnosis and Management
Treatment
Acute/Emergency:
- Peritoneal dialysis or hemodialysis (continuous renal replacement therapy) in neonates with severe toxic levels - rapidly clears BCAAs
- High-calorie, BCAA-free IV nutrition to halt catabolism
- Glucose infusion to suppress muscle protein breakdown
Chronic dietary management:
- BCAA-free synthetic formula - the cornerstone; provides all other amino acids, calories, and nutrients
- Controlled supplementation of leucine, isoleucine, and valine in amounts sufficient for growth but below toxic levels
- Leucine levels are monitored most closely (primary neurotoxin)
- During illness or catabolism, protein restriction must be intensified as endogenous muscle breakdown raises BCAA levels dangerously
Thiamine (B1) supplementation:
- High-dose thiamine in thiamine-responsive MSUD variants (stabilizes the BCKD complex)
Liver transplantation:
- Increasingly used - the liver is the primary site of BCAA oxidation
- Restores sufficient BCKD activity to relax dietary restrictions significantly
- A 2023 review notes benefits in metabolic stabilization but highlights risks and long-term challenges (Deon et al., Int J Dev Neurosci 2023)
Key point: BCAAs are the primary energy source during metabolic stress. Individuals with MSUD are at risk of acute metabolic decompensation during any catabolic state (infection, surgery, fasting).
- Lippincott Biochemistry 8th ed., p. 768-769
- Tietz Textbook of Laboratory Medicine 7th ed.
Neurologic Consequences
- Cerebral edema is the direct result of leucine accumulation in the brain
- Delayed diagnosis or poor control leads to intellectual disability, seizure disorder, spastic diplegia
- White matter abnormalities visible on diffusion-weighted MRI during acute decompensation (restricted diffusion in deep white matter, brainstem, cerebellum)
- Early diagnosis and strict metabolic control give the best neurocognitive outcome
A recent meta-analysis (Scharre et al., Genet Med 2025 - PMID 39431354) confirms that early diagnosis, disease variant, and quality of metabolic care are the strongest predictors of neurocognitive outcome.
Summary Table
| Feature | Detail |
|---|---|
| Inheritance | Autosomal recessive |
| Enzyme defect | BCKD complex (E1α, E1β, E2, or E3 subunit) |
| Substrate accumulating | Leucine, isoleucine, valine + keto acids |
| Pathognomonic marker | L-alloisoleucine in plasma |
| Odor | Maple syrup / caramelized sugar (urine, cerumen) |
| Key screening test | Newborn MS/MS screen; DNPH urine test |
| Confirmatory test | Plasma amino acids (elevated BCAAs + L-alloisoleucine) |
| Primary neurotoxin | Leucine (cerebral edema) |
| Odor source | Isoleucine keto acid (2-keto-3-methylvaleric acid) |
| Treatment | BCAA-restricted diet; thiamine in responsive variants; liver transplant |
| Outcome without treatment | Death within weeks (classic); intellectual disability in survivors |
Flashcard of it
Generating Image
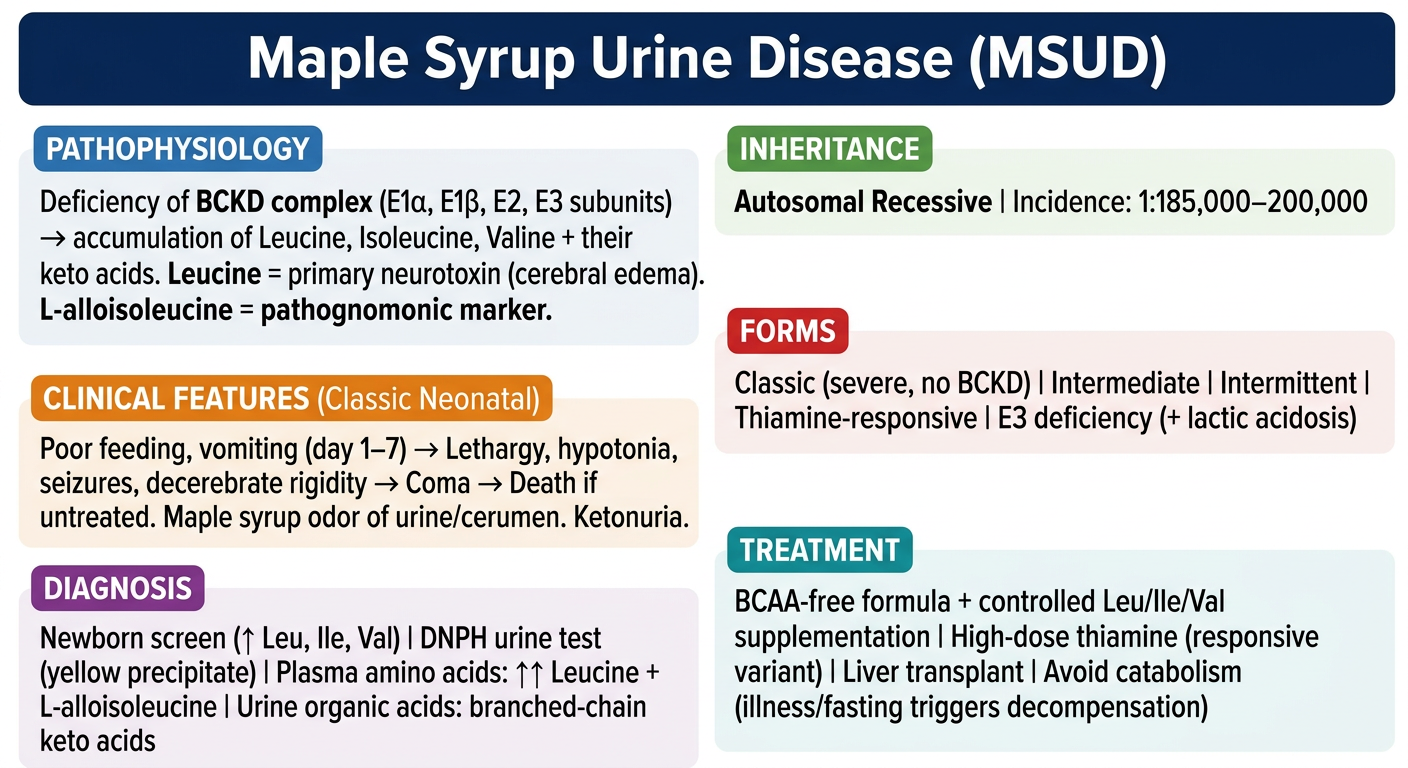
Here's your MSUD flashcard covering all the high-yield points: enzyme defect, inheritance, classic neonatal presentation, disease forms, diagnostic tests (including the pathognomonic L-alloisoleucine), and treatment principles. Great for quick review before exams.
This is a shared conversation. Sign in to Orris to start your own chat.