Case of a 20yr old with Aki post renal with hdun with renal stones with hypokalemia with severe hypocalcemia with Elevated phosphate with metabolic acidosis with pth Elevated 400 with 24hr urinary calcium 100mg with normal sized kidneys on usg
distal renal tubular acidosis type 1 nephrolithiasis hypokalemia hypocalcemia PTH elevated low urinary calcium
Clinical Analysis: 20-Year-Old with Post-Renal AKI + Hydronephrosis + Nephrolithiasis + Electrolyte Disturbances
🔑 Most Likely Unifying Diagnosis: Distal Renal Tubular Acidosis (Type 1 dRTA) → Causing Obstructive Nephropathy → Post-Renal AKI
Step-by-Step Pathophysiology
1. Primary Defect: Distal RTA (Type 1)
- Inability to acidify urine (urine pH persistently > 5.5 even during systemic acidosis)
- Normal anion gap hyperchloremic metabolic acidosis
- Hypokalemia — failure of the H⁺/K⁺-ATPase in the distal tubule means K⁺ is wasted renally regardless of how low serum K⁺ falls
2. Chronic Metabolic Acidosis → Bone Dissolution → Hypercalciuria
- Increased proximal citrate reabsorption (because intracellular acidosis consumes citrate) → hypocitraturia
- Citrate normally keeps calcium in solution; when it falls, CaHPO₄ and calcium phosphate precipitate
- Alkaline urine (pH > 6.5–7.0) further favors calcium phosphate stone formation
3. Bilateral Stones → Post-Renal AKI + Hydronephrosis
- Elevated BUN and creatinine (HDUN — high-degree urea nitrogen) — post-renal pattern
- Hydronephrosis on USG with normal kidney size (acute obstruction — kidneys haven't yet shrunk)
- Normal kidney size confirms this is acute, not chronic CKD
4. Secondary Hyperparathyroidism (PTH 400 pg/mL)
- Reduced GFR → phosphate retention → hyperphosphatemia ✓
- Hyperphosphatemia → depresses ionized calcium directly
- Reduced renal 1α-hydroxylase activity → ↓ calcitriol → less intestinal calcium absorption → hypocalcemia ✓
- Low calcium + high phosphate → PTH secretion markedly elevated (400 pg/mL) ✓
- This is secondary hyperparathyroidism (reactive, not autonomous)
"Phosphate retention leads to hyperparathyroidism by decreasing serum free calcium, which stimulates PTH secretion... A new steady state is achieved in which serum phosphate is restored to normal at the expense of a sustained high level of PTH." — Comprehensive Clinical Nephrology, 7th Ed.
5. Why is 24-Hour Urinary Calcium Only 100 mg (Low-Normal/Low)?
| Condition | Urinary Ca²⁺ | Explanation |
|---|---|---|
| Primary hyperparathyroidism | HIGH (>300 mg/day) | PTH promotes tubular Ca reabsorption but Ca filtered load is high |
| dRTA (baseline) | HIGH | Acidosis mobilizes bone Ca |
| dRTA with AKI/obstruction | LOW-NORMAL | Reduced GFR → less filtered Ca; PTH increases tubular reabsorption |
| Vitamin D deficiency | LOW | Poor intestinal absorption |
Complete Differential for the Underlying Cause of dRTA in a 20-Year-Old
| Cause | Clues to Look For |
|---|---|
| Primary/idiopathic dRTA | Most common in young females |
| Sjögren syndrome | Dry eyes/mouth, anti-Ro/La antibodies |
| SLE | ANA, complement levels, young female |
| Sickle cell anemia | CBC, Hb electrophoresis |
| Wilson disease | Liver function, ceruloplasmin, Kayser-Fleischer rings |
| Autosomal recessive (ATP6V1B1/ATP6V0A4 mutations) | Sensorineural deafness |
| Hypercalciuria/nephrocalcinosis causing dRTA | The stone disease itself can perpetuate tubular damage |
Investigations Needed
Confirm dRTA:
- Urine pH — should be > 5.5 even during acidosis
- Serum chloride — elevated (hyperchloremic acidosis, normal anion gap)
- Urine anion gap (Na⁺ + K⁺ − Cl⁻) — positive value confirms renal acid excretion failure
- ABG — degree of acidosis, respiratory compensation
- Urine citrate — will be very low in dRTA
For secondary HPT / metabolic bone disease:
- 25-OH Vitamin D — likely low
- 1,25(OH)₂D (calcitriol) — reduced due to impaired 1α-hydroxylase
- Serum calcium (total + ionized) — severe hypocalcemia
- Alkaline phosphatase — elevated if bone disease present
Imaging:
- Non-contrast CT KUB — better characterization of stones (calcium phosphate are visible)
- USG kidneys — already done (normal size, HDN confirmed)
- Skeletal survey / DEXA if osteitis fibrosa/osteomalacia suspected
For underlying cause of dRTA:
- ANA, anti-dsDNA, complement (SLE)
- Anti-SSA/anti-SSB (Sjögren)
- Hb electrophoresis (sickle cell)
- Serum ceruloplasmin, 24h urine copper (Wilson)
- Audiometry (genetic dRTA with deafness)
Management
Acute (Treat AKI first):
- Relieve obstruction — urological intervention: DJ stent or nephrostomy tube for bilateral HDN causing AKI
- IV fluids — careful correction of volume status (expect post-obstructive diuresis after relief)
- IV calcium gluconate — for severe symptomatic hypocalcemia (check for tetany, Chvostek/Trousseau signs, prolonged QTc on ECG)
- IV/oral potassium replacement — cautiously, monitor ECG
Chronic (Treat underlying dRTA):
- Potassium citrate (1–2 mEq/kg/day) — corrects acidosis AND hypokalemia AND hypocitraturia simultaneously; reduces stone formation
- Preferred over sodium bicarbonate when stones are present (sodium increases calciuria)
- Active Vitamin D (calcitriol 0.25–0.5 mcg/day) — once obstruction relieved, to correct hypocalcemia and suppress PTH
- Phosphate binders if phosphate persistently elevated after AKI resolution
- Treat underlying cause once identified (e.g., hydroxychloroquine for Sjögren/SLE)
Expected Outcomes After Treatment:
- PTH should normalize as hypocalcemia and hyperphosphatemia correct
- Urinary calcium will rise transiently post-AKI resolution (monitor for nephrocalcinosis)
- Continued alkali therapy prevents future stone formation
Summary Diagram of the Pathophysiology
dRTA (Type 1)
│
├── Metabolic acidosis (normal anion gap, hyperchloremic)
│ └── Bone buffering → Hypercalciuria + Hypocitraturia
│ └── Alkaline urine (pH >6.5)
│ └── Ca-phosphate STONES
│
├── Hypokalemia (H⁺/K⁺ ATPase failure → K⁺ wasting)
│
└── Bilateral stones → OBSTRUCTION → Post-renal AKI + HDN
│
└── Reduced GFR → ↑ Phosphate → ↓ Calcitriol
└── Hypocalcemia + Hyperphosphatemia
└── PTH 400 pg/mL (2° HPT)
└── Tubular Ca reabsorption ↑
└── Urinary Ca = 100 mg (low)
Key Teaching Points for This Case
- Normal anion gap metabolic acidosis + hypokalemia + stones in a young patient = dRTA until proven otherwise
- Low/normal urinary calcium in the context of AKI does NOT rule out hypercalciuric stone disease — the AKI itself suppresses urinary calcium
- PTH 400 pg/mL is secondary — it is a response to hypocalcemia + hyperphosphatemia from AKI, not primary hyperparathyroidism (which would cause HYPERcalcemia)
- Normal kidney size confirms acute obstruction (chronic would show cortical thinning)
- Potassium citrate is the cornerstone treatment for dRTA with stones — corrects acidosis, hypokalemia, and hypocitraturia in one agent
Hanging management
hanging near-hanging management emergency clinical guidelines airway cervical spine
hanging near-hanging management airway cervical spine cerebral edema complications
near hanging injury pathophysiology types judicial incomplete complete vascular airway spinal
hanging management airway intubation cervical spine neuroprotection ICP complications prognosis
Hanging / Near-Hanging: Clinical Management
Definitions & Classification
| Term | Definition |
|---|---|
| Hanging | Death from strangulation with body suspended by neck (own body weight) |
| Near-hanging | Survived a hanging attempt (reached hospital alive) |
| Complete hanging | Feet fully off ground — full body weight on neck |
| Incomplete hanging | Part of body still in contact with ground (most suicide attempts) |
| Judicial hanging | Drop ≥ victim's height — classic "hangman's fracture" |
Most ED presentations are incomplete near-hangings — very different pathophysiology from judicial hanging.
Pathophysiology of Injury
Primary Mechanisms (in order of clinical importance)
- Jugular veins compress first (low pressure) → venous outflow obstruction → stagnant cerebral hypoxia → loss of consciousness in 15 seconds
- Loss of muscle tone allows ligature to tighten → arterial occlusion (carotid/vertebral) → brain ischemia
- Carotid body compression → vagal reflex → bradycardia/cardiac arrest
- Carotid intimal tear → arterial dissection (delayed stroke risk)
- Requires more compressive force than veins
- Thyroid cartilage most commonly fractured
- Laryngotracheal disruption → subcutaneous emphysema
- Retropharyngeal/paratracheal hematoma → delayed airway compromise hours later
- Significant airway edema can develop over 12–24 hours
- Neurogenic pulmonary edema: massive sympathetic discharge from CNS injury → pulmonary vasoconstriction → capillary leak → ARDS
- Post-obstructive pulmonary edema: forceful inspiratory effort against obstruction → extreme negative intrapleural pressure → rapid-onset pulmonary edema on release
- Judicial/complete hanging: near-universal C-spine fracture (C2 "hangman's fracture"), cord transection
- Incomplete/near-hanging: C-spine fracture incidence < 1–5%; however, assume unstable until proven otherwise
- Mechanism: hyperextension + axial traction
Prehospital Management
- Immediately cut/remove ligature — support the body while cutting (don't allow sudden drop)
- Inline cervical spine immobilization — hard collar, log-roll technique
- Airway assessment — intubate only if airway acutely compromised; don't attempt ETI in the field without C-spine stabilization
- Basic life support / CPR if pulseless
- Bring ligature material to hospital (helps assess type/force)
Emergency Department Management
Primary Survey (ATLS Framework)
A — Airway (Highest Priority; Treat as PREDICTED DIFFICULT AIRWAY)
- Stridor, respiratory distress, hypoxia
- GCS ≤ 8 / unconscious
- Subcutaneous emphysema or suspected laryngotracheal injury
- Agitation/combativeness (cannot protect airway)
- Pre-oxygenate fully
- RSI with inline cervical immobilization — preferred
- Video laryngoscopy (hyperangulated) — first choice, especially with stridor
- Awake fiberoptic if time allows in a cooperative patient with stridor
- Have surgical airway immediately ready (scalpel + finger + bougie / cricothyroidotomy kit) — mark the cricothyroid membrane before beginning
- Ketamine is a useful induction agent (maintains hemodynamics, bronchodilation)
- If ETI fails → cricothyroidotomy → percutaneous trans-laryngeal ventilation as bridge
- Lung-protective strategy: Vt 6 mL/kg IBW, PEEP 5–8 cm H₂O
- PEEP also beneficial for post-obstructive pulmonary edema
- Target SpO₂ > 94%, avoid hypercapnia (worsens ICP)
B — Breathing
- High-flow O₂ for all patients
- CXR immediately — look for pulmonary edema, pneumothorax, pneumomediastinum
- Non-intubated patients with pulmonary edema → CPAP/BiPAP (PEEP)
C — Circulation
- Judicious fluids — avoid large volumes (exacerbates ARDS + cerebral edema)
- If hypotensive → vasopressors early rather than fluid loading
- Monitor for cardiac arrhythmias (continuous telemetry)
- 12-lead ECG — arrhythmias, QT prolongation from hypoxia
D — Disability (Neurological)
- GCS, pupils, motor/sensory exam
- Altered/comatose patient = assume raised ICP until proven otherwise
- Neuroprotective measures (see below)
E — Exposure
- Full skin exam — ligature mark location, petechiae (face/conjunctiva = venous obstruction), ecchymosis
- Look for co-ingestions (medication bottles at scene)
Investigations
| Investigation | Rationale |
|---|---|
| CT Brain | Cerebral edema, hemorrhage, anoxic injury |
| CT C-spine | Fracture/dislocation (all cases until cleared) |
| CTA Head/Neck | Carotid/vertebral artery dissection (critical — can cause delayed stroke) |
| Non-contrast CT neck | Laryngotracheal fracture, hematoma, subcutaneous emphysema |
| CXR | Pulmonary edema, pneumothorax, pneumomediastinum |
| ABG | Oxygenation, ventilation, metabolic status |
| ECG | Arrhythmias, QTc |
| CBC, CMP, coagulation | Baseline labs, toxicology screen |
| Soft tissue neck XR | If CT not immediately available |
Specific Complications and Management
1. Raised ICP / Cerebral Edema
- Head of bed 30° (neutral position, not rotation)
- Target SpO₂ > 94%, PaCO₂ 35–40 mmHg (avoid hypo- and hypercapnia)
- Avoid hyperthermia — aggressive fever control
- Therapeutic hypothermia — consider in post-cardiac arrest from hanging (32–36°C for 24 h; evidence extrapolated from cardiac arrest protocols)
- Seizure prophylaxis (levetiracetam or valproate)
- Mannitol / hypertonic saline if herniation signs
2. Carotid/Vertebral Artery Dissection
- Can be clinically silent initially → delayed stroke hours to days later
- CTA head/neck mandatory in all near-hanging victims
- If dissection confirmed: anticoagulation vs. antiplatelet therapy (individualized, neurosurgery/neurology input)
3. Pulmonary Edema / ARDS
- Lung-protective ventilation (LPV)
- PEEP titration
- Diuretics cautiously in neurogenic pulmonary edema
- ENT/bronchoscopy for laryngotracheal injury, dysphonia, stridor
4. Laryngotracheal Injury
- Symptoms: dyspnea, dysphonia, aphonia, odynophagia, subcutaneous emphysema
- ENT consultation + laryngobronchoscopy
- Monitor for tracheal stenosis as late complication
Disposition
| Clinical Status | Action |
|---|---|
| Cardiac arrest / coma | ICU, intubated, full critical care |
| Altered consciousness / any symptoms | Admit, continuous monitoring, repeat imaging |
| Asymptomatic with normal imaging | Observe minimum 24 hours — delayed airway/neurologic deterioration is well-documented |
| Discharge (only if truly asymptomatic after 24 h) | Mandatory psychiatric consultation first; strict return precautions |
⚠️ Do NOT discharge any near-hanging victim without psychiatric evaluation. All intentional cases require psychiatry/crisis team consultation before disposition.
Prognosis
| Finding | Implication |
|---|---|
| Brief suspension, rapid rescue, GCS normal | Generally good prognosis |
| Prolonged suspension / absent pulse at scene | High mortality; severe anoxic brain injury likely |
| Neurogenic pulmonary edema | Poor prognostic sign |
| Coma on arrival | ~50% mortality or severe neurologic disability |
| Judicial hanging reaching ED | Near-universal severe cord injury |
Summary: Key Principles
- Vascular compression, not airway obstruction, causes the primary brain injury — act fast
- Treat as a difficult airway — have surgical backup ready before starting
- Judicious fluids — vasopressors over volume loading
- CTA neck in every case — don't miss carotid dissection
- 24-hour minimum observation even for "well-looking" patients
- Neurogenic and post-obstructive pulmonary edema are major causes of delayed death
- Psychiatric consultation is mandatory in all intentional near-hangings
In the above discussed case what if the bp is high
Adding Hypertension to This Case: What It Changes
- Post-renal AKI + hydronephrosis + nephrolithiasis
- Hypokalemia + metabolic acidosis (normal anion gap)
- Severe hypocalcemia + hyperphosphatemia + PTH 400
- Low urinary calcium (100 mg/day)
- Normal kidney size on USG
Why This Combination Is Diagnostically Critical
dRTA (Type 1) does NOT cause hypertension — it is a normotensive or hypotensive condition.
Causes of Hypertension in This Setting
1. Post-Renal AKI Itself → Fluid Retention and RAAS Activation
- Bilateral obstruction reduces GFR to near-zero → inability to excrete sodium and water → volume overload → hypertension
- Reduced renal perfusion activates juxtaglomerular cells → ↑ Renin → ↑ Angiotensin II → ↑ Aldosterone → further sodium retention and BP elevation
- This is a secondary/renovascular-type hypertension mediated by obstruction
2. Secondary Hyperparathyroidism / PTH-Mediated Hypertension
- PTH stimulates vascular smooth muscle calcium entry → increased peripheral vascular resistance
- Calcitriol deficiency impairs endothelium-dependent vasodilation
- This is a well-recognized association in CKD-MBD (chronic kidney disease–mineral bone disorder)
3. ⚠️ The Game-Changer: Hypertension + Hypokalemia = Rule Out Secondary Hyperaldosteronism
"The coexistence of hypertension and spontaneous hypokalemia should always raise the possibility of secondary causes of hypertension."
A. Primary Aldosteronism (Conn Syndrome)
- High aldosterone + suppressed renin → Aldosterone:Renin ratio (ARR) > 30
- Causes: adrenal adenoma (Conn) or bilateral adrenal hyperplasia
- Hypokalemia ✓, Hypertension ✓
- BUT: Primary aldosteronism causes metabolic ALKALOSIS, not acidosis
- And it causes hyperkalemic correction — urinary K wasting with alkalosis
- Does NOT fit metabolic acidosis in this case
B. Secondary Hyperaldosteronism (Renin-driven)
- High aldosterone + high renin → ARR < 20
- Caused here by: bilateral obstructive AKI → reduced renal perfusion → juxtaglomerular renin release
- Hypokalemia ✓, Hypertension ✓
- Metabolic alkalosis expected (aldosterone drives H⁺ secretion) — BUT in this case, the underlying dRTA + AKI produces acidosis that overrides this alkalotic tendency
- This is likely the dominant mechanism of hypertension here
C. Liddle Syndrome (Monogenic — important in a 20-year-old!)
- Gain-of-function mutation in ENaC (epithelial sodium channel) → constitutive sodium retention
- Hypertension + hypokalemia + low aldosterone + low renin
- Metabolic alkalosis (not acidosis) → less likely to be primary cause here
- But worth excluding given young age
4. The Most Important New Differential to Consider: Gordon Syndrome (Pseudohypoaldosteronism Type 2 / Familial Hyperkalemic Hypertension)
5. Reframe: Could This Be a Completely Different Unifying Diagnosis?
Distal RTA Secondary to Sjögren Syndrome / SLE
- Autoimmune tubular injury → dRTA
- Same diseases cause immune-complex glomerulonephritis → reduced GFR → hypertension
- ANA, anti-SSA/SSB, complement levels become even more critical to check now
Primary Hyperparathyroidism (Revisit)
- PTH 400 with hypocalcemia = secondary HPT ✓
- But if serum calcium were HIGH (not low), primary HPT would cause:
- Hypercalcemia → hypertension
- Nephrolithiasis ✓
- Metabolic acidosis (proximal RTA from PTH effect on proximal tubule)
- Urinary calcium would be HIGH, not 100 mg
- Excluded by hypocalcemia in this case — PTH is reactive, not autonomous
Updated Investigations to Add
| Test | Purpose |
|---|---|
| Plasma Renin Activity (PRA) | High in secondary hyperaldosteronism from obstruction |
| Plasma Aldosterone | Elevated if secondary hyperaldosteronism |
| Aldosterone:Renin Ratio (ARR) | >30 suggests primary aldosteronism |
| Urine sodium, potassium, chloride | Assess tubular handling |
| TTKG (transtubular K gradient) | >2 in hypokalemia = renal K wasting confirmed |
| Renal Doppler USG | Renal artery stenosis as cause? (less likely bilaterally in a 20-year-old) |
| ANA, anti-dsDNA, complement, anti-SSA/SSB | Autoimmune cause of both dRTA and glomerulonephritis |
| Echo / fundoscopy | Target organ damage from hypertension |
| 24h urine catecholamines/metanephrines | Phaeochromocytoma (if headache, sweating, palpitations) |
Antihypertensive Management: Drug Choice Matters
| Drug | Use / Avoid | Reason |
|---|---|---|
| ACE inhibitor / ARB | ⚠️ Use with extreme caution / avoid acutely | Post-renal AKI — bilateral obstruction → reducing angiotensin II can precipitate haemodynamic AKI; also risk of hyperkalaemia (though here K is low) |
| Calcium channel blocker (amlodipine) | ✅ Good choice | Lowers BP, no renal tubular effects, benefits vascular smooth muscle (excess PTH effect) |
| Loop diuretic (furosemide) | ✅ If volume overload confirmed | Treats volume-mediated hypertension; also aids phosphate excretion |
| Beta-blocker | ✅ Adjunct | Suppresses renin release if renin-driven |
| Thiazide diuretic | ❌ Avoid | Worsens hypokalemia; hypocalciuria (blocks calcium excretion — counterproductive here) |
| Spironolactone | ⚠️ Consider only if primary aldosteronism confirmed | Risk of hyperkalaemia in AKI |
| Potassium citrate | ✅ Essential | Corrects acidosis AND hypokalemia — do not use potassium chloride alone |
⚠️ Do not start ACE inhibitor/ARB until obstruction is relieved and GFR is recovering. In post-renal AKI, RAAS blockade can drop GFR precipitously.
Updated Unifying Pathophysiology with Hypertension
dRTA (Type 1) — Primary Defect
│
├── Metabolic acidosis + hypokalemia + alkaline urine
│ └── Calcium phosphate stones (bilateral)
│ └── Bilateral obstruction
│ ├── Post-renal AKI (BUN↑, creatinine↑)
│ │ └── Fluid retention → HYPERTENSION ✓
│ └── Reduced renal perfusion
│ └── ↑ Renin → ↑ Angiotensin II → ↑ Aldosterone
│ └── More Na retention → MORE HYPERTENSION ✓
│ └── (Aldosterone-driven K wasting adds to hypokalemia)
│
└── Reduced GFR → ↑ Phosphate → ↓ Calcitriol → Hypocalcemia → PTH 400 (2° HPT)
└── PTH → vascular SMC Ca entry → HYPERTENSION ✓
Key Teaching Points
- Hypertension in post-renal AKI = volume overload + RAAS activation from reduced renal perfusion — it is expected and should resolve after relieving obstruction
- Hypertension + hypokalemia together mandate a renin/aldosterone pair to distinguish primary from secondary hyperaldosteronism
- Metabolic acidosis excludes primary aldosteronism as the sole cause (which causes alkalosis)
- In a 20-year-old, always think autoimmune cause (Sjögren/SLE) — it can cause dRTA, glomerulonephritis, and hypertension simultaneously
- Drug of choice for BP: calcium channel blocker acutely; avoid ACE inhibitor/ARB until obstruction resolved and GFR recovering
- If BP does NOT normalise after obstruction is relieved → mandatory full secondary hypertension workup
Wernickes encephalopathy and pure upbeat nystagmus
pure upbeat nystagmus Wernicke encephalopathy mechanism pontomesencephalic ventral tegmental tract thiamine
upbeat nystagmus Wernicke encephalopathy mechanism pontomesencephalic lesion
Wernicke's Encephalopathy & Pure Upbeat Nystagmus
1. Wernicke's Encephalopathy — Overview
Causes / Risk Factors
| Primary Cause | Secondary/Non-Alcoholic Causes |
|---|---|
| Chronic alcoholism (most common) | Hyperemesis gravidarum |
| Poor dietary intake | Malignancy / chemotherapy |
| Impaired GI absorption | Bariatric surgery / gastric bypass |
| Increased thiamine demand (glucose loading) | Prolonged TPN without B vitamins |
| Hemodialysis (water-soluble vitamin loss) | Crohn's disease, malabsorption |
| HIV/AIDS, renal dialysis |
Body thiamine stores last only ~18 days — deficiency develops rapidly in vulnerable states.
2. Pathophysiology
| Enzyme | Pathway | Consequence of Deficiency |
|---|---|---|
| Pyruvate dehydrogenase | Krebs cycle entry | Pyruvate/lactate accumulation |
| α-Ketoglutarate dehydrogenase | Krebs cycle | Glutamate accumulation → excitotoxicity |
| Transketolase | Pentose phosphate pathway | Reduced NADPH → oxidative stress |
3. Classic Triad — and Why It's Rarely Complete
- Ophthalmoplegia / Ocular abnormalities
- Cerebellar ataxia (gait/truncal)
- Global confusion / Encephalopathy
⚠️ The full triad is present in only ~30% of patients. Up to 80% are missed clinically and diagnosed only at autopsy.
- Ataxia
- Ophthalmoplegia / Nystagmus
- Confusion / Memory disturbance
- Hypothermia + hypotension
- Unconsciousness
4. Ocular Abnormalities in Wernicke's Encephalopathy
| Finding | Mechanism | Notes |
|---|---|---|
| Horizontal nystagmus on lateral gaze | Most common; vestibular nucleus involvement | Classically described |
| Lateral rectus palsy (VI nerve) | Abducens nucleus, often bilateral | Diplopia ± esotropia |
| Conjugate gaze palsies | MLF / PPRF dysfunction | Internuclear ophthalmoplegia |
| Vertical nystagmus (upbeat or downbeat) | Pontomesencephalic / vestibular nuclei | See below |
| Ptosis | Rare; III nerve nucleus | Advanced disease |
| Miosis | Autonomic involvement | Late finding |
"Ocular motor abnormalities include horizontal nystagmus on lateral gaze, lateral rectus palsy (usually bilateral), conjugate gaze palsies, and rarely ptosis." — Harrison's Principles of Internal Medicine 22e
5. Pure Upbeat Nystagmus in Wernicke's Encephalopathy
What is Pure Upbeat Nystagmus?
- Present in primary position (not just gaze-evoked)
- Increases on upgaze, may decrease on downgaze
- Typically bilateral and conjugate
Localization — The Key Teaching Point
| Nystagmus Type | Anatomical Localization |
|---|---|
| Downbeat nystagmus | Bilateral cervicomedullary junction (flocculus) |
| Upbeat nystagmus | Bilateral pontomesencephalic junction |
| Bow-tie nystagmus (variant of upbeat) | Bilateral pontomedullary junction / cerebellar vermis |
| Periodic alternating nystagmus | Floor of 4th ventricle |
Why Does Wernicke's Cause Upbeat Nystagmus Specifically?
-
Periaqueductal grey matter (PAG) — damaged by thiamine deficiency; PAG integrates vertical gaze signals from the interstitial nucleus of Cajal (INC) and rostral interstitial nucleus of the MLF (riMLF)
-
Ventral tegmental tract — fibers from the anterior semicircular canals (which encode upward head movement) ascend via this pathway at the pontomesencephalic junction; damage biases the vertical vestibulo-ocular reflex toward an upward fast phase
-
Cerebellar anterior vermis — normally provides inhibitory input suppressing upward drift; thiamine-dependent neurons in the vermis fail → disinhibition → eyes drift down → corrective upbeat fast phase
-
Medial vestibular nuclei — these nuclei contain thiamine-sensitive neurons; selective bilateral dysfunction here generates tonic upbeat drift
- Periaqueductal grey involvement (characteristic WE lesion site)
- Superior vestibular nucleus dysfunction
- Anterior vermis involvement
Upbeat vs. Horizontal Nystagmus in WE
| Feature | Horizontal Nystagmus | Upbeat Nystagmus |
|---|---|---|
| Frequency in WE | More common (~classic) | Less common (~40% in dedicated series) |
| Mechanism | Lateral vestibular nucleus | Pontomesencephalic / anterior vermis |
| MRI correlate | Medial thalami, periaqueductal grey | Periaqueductal grey, pontine tegmentum |
| Response to thiamine | Usually resolves | Usually resolves; may persist |
| Horizontal nystagmus persists | Yes — may be residual | — |
6. Pathological Anatomy & MRI Findings
Periventricular regions:
• Medial thalami (dorsomedial nuclei) ← amnestic defect
• Mammillary bodies ← most characteristic gross finding
• Periaqueductal grey matter (midbrain) ← ophthalmoplegia
• Floor of 4th ventricle / pontine tegmentum ← nystagmus
• Walls of 3rd ventricle
• Cerebellar vermis (anterior)
- T2/FLAIR hyperintensity — bilateral, symmetrical in medial thalami, mammillary bodies, periaqueductal grey, 3rd ventricular walls, pons, medulla
- DWI restriction — cytotoxic oedema in acute phase
- T1 post-contrast enhancement of mammillary bodies — hallmark of acute WE
- Microbleeds on GRE/SWI in thalami and mammillary bodies = poor prognostic sign
- Cortical involvement — rare, indicates severe/atypical WE
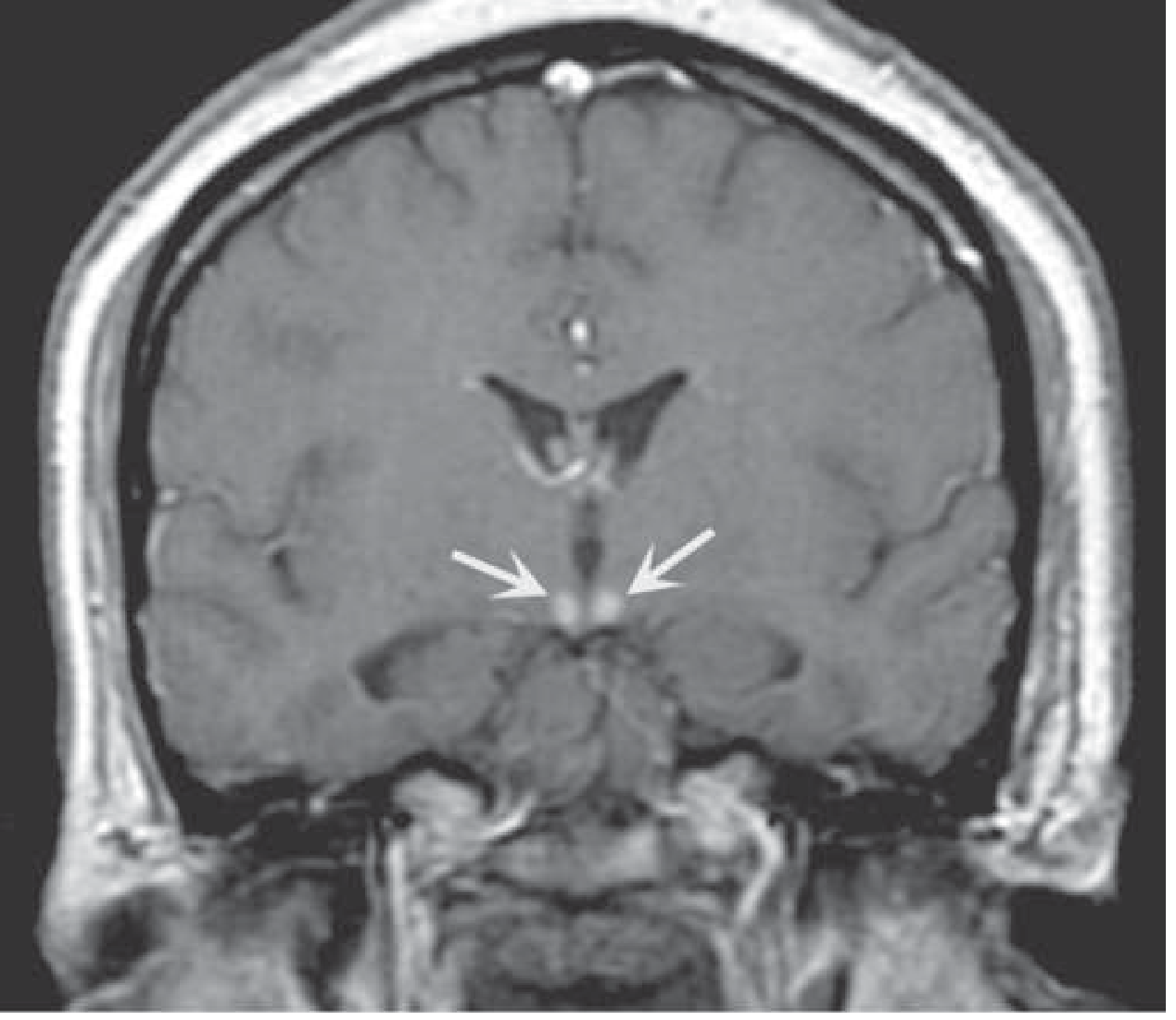
7. Differential Diagnosis of Pure Upbeat Nystagmus
| Cause | Mechanism |
|---|---|
| Wernicke's encephalopathy | Pontomesencephalic thiamine-sensitive nuclei |
| Brainstem glioma | Pontine tegmentum / PAG |
| MS (demyelination) | MLF / pontomesencephalic plaques |
| Cerebellar anterior vermis lesion | Disinhibition of upward drift |
| Medullary infarction | Nucleus prepositus hypoglossi / medial vestibular nucleus |
| Meningitis / encephalitis | Brainstem inflammation |
| Drug toxicity | Baclofen, carbamazepine, anticonvulsants |
| Organophosphate poisoning | Nicotinic receptor effects |
| Tobacco smoking | Nicotine effect on brainstem |
In the right clinical context (malnourishment, alcohol use, vomiting), pure upbeat nystagmus should immediately trigger empiric thiamine administration — do not wait for imaging or labs.
8. Treatment — Medical Emergency
Principle: Give thiamine BEFORE glucose
Thiamine Dosing Protocol
| Setting | Dose | Route | Duration |
|---|---|---|---|
| Suspected WE (treatment) | 500 mg TID | IV (preferred) | 2–3 days |
| Ongoing treatment after acute phase | 250 mg OD | IV or IM | 5 more days |
| Maintenance | 100–300 mg OD | Oral (after IV course) | Long-term |
| Prophylaxis (at-risk, hospitalised) | 200–300 mg OD | IM or IV | 3–5 days |
| Prophylaxis (community, low-risk) | 300 mg OD | Oral | During withdrawal period |
⚠️ Oral thiamine is unreliable in alcoholics — impaired GI absorption means IV/IM is mandatory for treatment and high-risk prophylaxis.
Co-administer:
- Magnesium (magnesium is a cofactor required for thiamine phosphorylation — hypomagnesaemia blunts thiamine response)
- Other B vitamins (folate, B12, B6)
- Correct electrolytes, nutritional deficiencies
Expected Response to Thiamine:
| Feature | Response |
|---|---|
| Ophthalmoplegia / gaze palsies | Improves within hours |
| Upbeat/horizontal nystagmus | Improves in days; horizontal nystagmus may persist |
| Ataxia | Slower; improves over weeks; ~50% have residual ataxia |
| Confusion / encephalopathy | Gradual improvement over days |
| Memory defect (Korsakoff) | Often permanent — does not respond to thiamine |
9. Wernicke → Korsakoff Progression
- Amnestic defect becomes permanent
- Korsakoff psychosis: anterograde amnesia + confabulation + disordered temporal sequencing
- Pathological basis: lesions in dorsomedial thalamic nuclei (not mammillary bodies per se)
- Korsakoff amnesia responds poorly to thiamine — prevention is the only strategy
Key Teaching Points
- Upbeat nystagmus in WE localises to the bilateral pontomesencephalic junction — the same periventricular zone that thiamine deficiency preferentially injures
- Nystagmus in WE is most commonly horizontal (classic), but vertical upbeat nystagmus occurs in ~40% and is equally valid as a diagnostic sign
- The triad is present in only 30% — treat empirically on any single sign in a susceptible patient
- Give 500 mg IV thiamine TID BEFORE any glucose — this is the single most important clinical rule
- Magnesium deficiency must be corrected alongside thiamine — without it, thiamine therapy is ineffective
- Ophthalmoplegia and nystagmus resolve quickly; Korsakoff amnesia is irreversible — early treatment is the only prevention
- MRI can be normal in up to 50% of acute WE — a negative MRI does not rule out WE
SCA With epilepsy
spinocerebellar ataxia with epilepsy SCA types seizures SCA10 SCA17 DRPLA mechanism antiepileptic treatment
SCA with epilepsy antiepileptic drug choice cerebellar ataxia avoid worsen levetiracetam valproate
spinocerebellar ataxia epilepsy seizures SCA10 DRPLA
Spinocerebellar Ataxia (SCA) with Epilepsy
Overview
Classification of SCAs with Epilepsy
Three Broad Categories:
| Category | Examples | Mechanism |
|---|---|---|
| SCA subtypes where epilepsy is a defining feature | SCA10, SCA17, DRPLA | Repeat expansion causes neurodegeneration of epileptogenic circuits |
| SCAs where epilepsy occurs occasionally | SCA2, SCA3 | Cortical/subcortical involvement; modifier genes |
| Mitochondrial ataxia-epilepsy syndrome | SCAE (POLG-related, MEMSA) | mtDNA depletion; cerebellar + cortical energy failure |
1. SCA10 — The Prototypic "Ataxia + Epilepsy" SCA
Genetics
- Gene: ATXN10 (chromosome 22q13)
- Mutation: Expanded ATTCT pentanucleotide intronic repeat in intron 9 — not a trinucleotide CAG repeat
- Normal: 10–22 repeats; Pathological: 800–4500 repeats
- Mechanism: toxic RNA gain of function (not polyglutamine toxicity) → neurodegeneration via RNA-mediated mechanisms
Epidemiology
- Predominant in Latin American populations (Mexico, Brazil)
- Autosomal dominant; onset age 18–45 (mean ~35 years)
Clinical Features
| Feature | Details |
|---|---|
| Cerebellar ataxia | Gait ataxia, dysarthria, dysmetria — first symptom |
| Epilepsy | Appears years after ataxia onset |
| Seizure types | Generalized tonic-clonic (most common) AND complex partial seizures |
| Nystagmus | Coarse gaze-evoked; saccade velocity normal |
| Pyramidal signs | Mild hyperreflexia, Babinski (minority) |
| Peripheral neuropathy | Present in some families |
| Cognitive dysfunction | Mild (IQ ~70 in some); not frank dementia |
| Mood disorders | Depression common |
Epilepsy Frequency by Ethnicity (Critical Fact)
| Population | Epilepsy Frequency |
|---|---|
| Mexican ancestry | ~60–80% — very high |
| Brazilian (Paraná region) | ~65% |
| Brazilian (other regions) | ~7% |
| Other | Rare |
The striking inter-family variation in epilepsy frequency suggests modifier genes influence epilepsy expression independent of ATTCT repeat size (no correlation with repeat length).
Key Warning
SCA10 seizures can progress to status epilepticus, which may be fatal — seizure control is therefore a top management priority.
2. DRPLA (Dentatorubral-Pallidoluysian Atrophy)
Genetics
- Gene: ATN1 (chromosome 12p13)
- Mutation: CAG trinucleotide repeat expansion
- Normal: ≤35 repeats; Pathological: ≥49 repeats
- Marked anticipation (especially paternal transmission)
Epidemiology
- Highest frequency in Japan (~20x more common than in Caucasians)
- Mean age of onset: ~29 years (range 1–60)
Clinical Phenotype (Age-Dependent)
| Age of Onset | Dominant Features |
|---|---|
| Juvenile/early onset (<20 years) | Progressive myoclonic epilepsy (PME), myoclonus, cerebellar ataxia, dementia, mental retardation |
| Late onset (>40 years) | Cerebellar ataxia, choreoathetosis, dementia, psychiatric symptoms (resembles HD) |
DRPLA as Progressive Myoclonic Epilepsy
- Action myoclonus
- Stimulus-sensitive myoclonus
- Generalised tonic-clonic seizures
- Atonic/absence seizures
- EEG: polyspike-wave, photoparoxysmal response
- Progressive cerebellar ataxia + PME + dementia in a young person = DRPLA until proven otherwise (especially in Asian patients)
3. SCA17
Genetics
- Gene: TBP (TATA box-binding protein), chromosome 6q27
- Mutation: CAG/CAA repeat expansion (normal ≤42; pathological ≥47)
- Overlaps phenotypically with Huntington's disease
Clinical Features with Epilepsy
- Progressive cerebellar ataxia + psychiatric symptoms (psychosis, depression) + dementia
- Seizures — reported in a subset; PME has been described (rare)
- Nocturnal frontal lobe epilepsy also reported
- Longer CAG repeats → earlier onset, HD-like phenotype
- Shorter repeats → later onset, ataxia-predominant
4. SCAE — "Spinocerebellar Ataxia with Epilepsy" (POLG-Related / MEMSA)
Genetics
- Gene: POLG (polymerase gamma) — mitochondrial DNA polymerase
- Inheritance: Autosomal recessive
- Part of the POLG-related disorder spectrum (also includes Alpers syndrome, CPEO, MELAS-like, SANDO)
- Also called MEMSA (Myoclonic Epilepsy, Myopathy, Sensory Ataxia)
Clinical Features
| Feature | Details |
|---|---|
| Cerebellar ataxia | First symptom; onset young adulthood |
| Epilepsy | Develops after ataxia; typically begins in right arm → generalization |
| Myoclonus | Action and stimulus-sensitive |
| Sensory neuropathy | Present |
| Myopathy | Proximal or distal; exercise intolerance |
| Ptosis + external ophthalmoplegia | Late-onset |
| Progressive cognitive impairment | Dementia/encephalopathy |
| Liver failure | ⚠️ Can occur — especially precipitated by sodium valproate |
Critical Management Point
Sodium valproate is CONTRAINDICATED in POLG/SCAE — it can precipitate fatal hepatic failure by inhibiting mitochondrial function in already-compromised mtDNA replication.
5. SCA2 / SCA3 with Incidental Epilepsy
- These are occasional, not defining
- In SCA2 families, focal epilepsy has been reported and attributed to co-existing epilepsy susceptibility genes (modifier effect)
- SCA3 (Machado-Joseph disease): seizures are not a prominent feature but can occur with advanced disease
Diagnostic Approach
Ataxia + Epilepsy in same patient
│
├── Family history? Autosomal dominant? → SCA10, SCA17, DRPLA
│
├── Latin American ancestry? → SCA10 (especially)
│
├── Japanese/Asian ancestry? → DRPLA (especially)
│
├── PME phenotype (myoclonus + ataxia + seizures)?
│ ├── Young onset → DRPLA, POLG/SCAE
│ └── Also consider: MERRF, Lafora disease, Unverricht-Lundborg
│
├── Autosomal recessive + myopathy + neuropathy? → POLG/SCAE
│
└── Psychiatric + dementia + ataxia? → SCA17, DRPLA (late)
Investigations
- Genetic panel — SCA repeat expansions (SCA1–3, 6, 7, 10, 17, DRPLA); POLG sequencing
- MRI brain — cerebellar atrophy (vermis + hemispheres); in DRPLA also basal ganglia + brainstem atrophy
- EEG — characterise seizure type; polyspike-wave = PME pattern; photoparoxysmal response in DRPLA
- Nerve conduction / EMG — neuropathy (SCAE, SCA10)
- Muscle biopsy + mitochondrial studies — if POLG/SCAE suspected; ragged red fibres on modified Gomori trichrome
- Liver function tests — baseline before any AED; critical in POLG
Management of Epilepsy in SCA
The Critical Challenge: Many AEDs Worsen Ataxia
| AED | Risk of Worsening Ataxia | Comment |
|---|---|---|
| Phenytoin | ⚠️ HIGH (37.9%) | Cerebellar toxicity even at therapeutic levels; chronic use causes irreversible cerebellar atrophy |
| Carbamazepine / Oxcarbazepine | ⚠️ HIGH (OXC ~30%) | Cerebellar adverse effects common; use with caution |
| Clonazepam | ⚠️ HIGH (50%) | Sedation + ataxia severely limiting |
| Gabapentin / Pregabalin | MODERATE (9–10%) | Can worsen balance; occasional use in SCA6 for ataxia |
| Lamotrigine | MODERATE (18.5% in RCTs) | Dizziness and ataxia; start low |
| Topiramate | LOW–MODERATE (6.6%) | May worsen ataxia in SCA17 at >25–50 mg/day |
| Zonisamide | MODERATE (12.7%) | |
| Levetiracetam | ✅ LOW (~1.5%) | First-line preferred in SCA |
| Valproate | MODERATE (3.6%) | Useful in PME/DRPLA BUT absolutely contraindicated in POLG/SCAE |
| Sodium valproate | ❌ CONTRAINDICATED | In POLG/SCAE — risk of fatal hepatotoxicity |
Drug Recommendations by Subtype
| Condition | Preferred AED(s) | Avoid |
|---|---|---|
| SCA10 | Levetiracetam, valproate, carbamazepine | Phenytoin (chronic use) |
| DRPLA (PME) | Valproate ± clonazepam (for myoclonus), levetiracetam, zonisamide | Lamotrigine (can worsen myoclonus) |
| SCAE/POLG | Levetiracetam, lamotrigine, lacosamide | Valproate (ABSOLUTELY), phenytoin |
| SCA17 | Levetiracetam, lamotrigine | Topiramate >25 mg (worsened ataxia reported) |
General Principles
- Levetiracetam — drug of choice across most SCA-epilepsy syndromes (lowest cerebellar side-effect profile)
- Always screen for POLG mutation before prescribing valproate in any young ataxia-epilepsy patient
- Avoid phenytoin long-term — chronic cerebellar toxicity causes permanent worsening of ataxia; paradoxically most commonly used historically
- Clonazepam — useful for myoclonus in DRPLA but causes profound ataxia and sedation; use minimally
- Status epilepticus in SCA10 — treat aggressively (IV benzodiazepine → IV levetiracetam → anaesthesia if refractory); can be fatal
Ataxia Management (Symptomatic)
| Treatment | Evidence |
|---|---|
| Riluzole 50 mg BD | Class I evidence for improvement in mixed and SCA ataxia (SARA scores); best current option |
| Valproate (SCA3 only) | Possibly improves ataxia at 1200 mg/day in SCA3 (Class II) |
| Physiotherapy / gait training | Standard care; preserves function |
| Speech therapy | For dysarthria and dysphagia |
| Troriluzole | In expanded access program for all SCA types (FDA review pending) |
| Acetazolamide | Useful in episodic ataxia types EA1/EA2 (distinct from SCA) |
Summary Table: Key SCA-Epilepsy Subtypes
| Feature | SCA10 | DRPLA | SCAE (POLG) | SCA17 |
|---|---|---|---|---|
| Inheritance | AD | AD | AR | AD |
| Gene | ATXN10 | ATN1 | POLG | TBP |
| Mutation | ATTCT pentanucleotide | CAG | Point mutations | CAG/CAA |
| Epilepsy type | GTCS ± partial | PME (early), GTCS | PME + myoclonus | GTCS, PME (rare) |
| Epilepsy frequency | 60–80% (Mexico) | Juvenile onset = defining | Defining | Occasional |
| Key other feature | Latin American | Choreoathetosis / dementia | Hepatotoxicity with VPA | Psychiatric / HD-like |
| Anticonvulsant choice | LEV, VPA, CBZ | VPA + LEV | LEV (NOT VPA) | LEV, LTG |
Key Teaching Points
- SCA + epilepsy should immediately prompt genetic testing for SCA10 and DRPLA — these are the two main SCAs where epilepsy is a defining, frequent feature
- SCAE/POLG (mitochondrial) is the specific named entity "spinocerebellar ataxia with epilepsy" — distinct from polyglutamine SCAs; valproate is lethal
- Phenytoin must be avoided long-term in any ataxia patient — it causes cerebellar degeneration
- Levetiracetam has the lowest cerebellar side-effect burden and is the preferred first-line AED across all SCA-epilepsy syndromes
- DRPLA in a young Asian patient with PME + ataxia + dementia = classic triad; marked anticipation explains severe juvenile presentation in offspring of mildly affected parents
- In SCA10, epilepsy severity does not correlate with repeat size but does vary markedly between families — modifier genes are implicated
Hereditary causes of neuropathy presented from birth in the form of sensory ataxia
hereditary neuropathies presenting from birth sensory ataxia congenital onset HSAN Friedreich ataxia CMT types
hereditary sensory neuropathy congenital onset sensory ataxia HSAN CMT
hereditary sensory ataxic neuropathy early onset congenital CANVAS SANDO POLG
hereditary neuropathy sensory ataxia from birth congenital types HSAN Friedreich ataxia-telangiectasia Refsum CANVAS differential
hereditary sensory neuropathy congenital onset ataxia review
hereditary neuropathy sensory ataxia congenital neonatal HSAN types large fiber proprioception loss Friedreich onset age
"sensory ataxic neuropathy" hereditary early onset congenital differential diagnosis
Hereditary Causes of Neuropathy Presenting from Birth as Sensory Ataxia
Conceptual Framework
- Positive Romberg's sign (ataxia worse with eyes closed)
- Loss of joint position sense and vibration
- Absent deep tendon reflexes (areflexia)
- Normal or near-normal cerebellar examination (limb ataxia only when proprioceptive input is lost)
- Pseudoathetosis of fingers (unstable posture of outstretched hands)
- Dorsal root ganglion (DRG) neuron — neuronopathy/ganglionopathy
- Peripheral sensory axon — sensory neuropathy
- Posterior/dorsal columns — myelopathy
- Any combination of all three
Classification by Onset and Inheritance
I. Hereditary Sensory and Autonomic Neuropathies (HSAN)
HSAN Type I (Riley-Day Variant / Hereditary Sensory Neuropathy Type I)
| Feature | Details |
|---|---|
| Inheritance | Autosomal dominant |
| Gene | SPTLC1, SPTLC2, ATL1, DNMT1 |
| Onset | Late childhood / adolescence (NOT true congenital) |
| Predominant fibre loss | Large > small fibres |
| Sensory features | Proprioception loss, vibration loss, sensory ataxia |
| Autonomic | Minimal |
| Motor | Variable wasting of distal legs |
| Key complications | Charcot joints, painless ulcers, osteomyelitis |
HSAN I is the most common hereditary pure sensory neuropathy but presents in teens/young adults, not strictly at birth.
HSAN Type III — Familial Dysautonomia (Riley-Day Syndrome)
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | IKBKAP (chromosome 9q31) |
| Onset | Congenital / from birth ✓ |
| Ethnicity | Ashkenazi Jewish (carrier frequency ~1:30) |
| Predominant fibre loss | Small fibres predominantly, BUT large fibres also affected |
| Sensory features | Reduced pain/temperature; vibration relatively preserved; sensory ataxia develops with large fibre involvement |
| Autonomic features (major) | Absent lacrimation, orthostatic hypotension, episodic hypertension, vomiting crises, dysregulated temperature |
| Motor | Hypotonia, absent DTRs |
| Other | Absent fungiform papillae on tongue (pathognomonic), scoliosis |
Although primarily a small-fibre neuropathy, large fibre involvement produces sensory ataxia as a secondary feature in HSAN III.
HSAN Type II — Congenital Sensory Neuropathy
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | WNK1/HSN2, FAM134B, KIF1A |
| Onset | From birth / early infancy ✓ |
| Fibre loss | All sensory fibres (large + small) |
| Sensory features | Global sensory loss; proprioception and vibration lost → sensory ataxia |
| Autonomic | Variable |
| Key complications | Self-mutilation, recurrent painless injuries, Charcot joints |
| DTRs | Absent |
HSAN Type IV — Congenital Insensitivity to Pain with Anhidrosis (CIPA)
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | NTRK1 (TrkA — NGF receptor) |
| Onset | Congenital ✓ |
| Fibre loss | Small unmyelinated fibres (C fibres) — NOT large fibres |
| Sensory features | No pain, no temperature; vibration and proprioception PRESERVED |
| Result | No sensory ataxia — large fibres intact |
| Other | Anhidrosis, intellectual disability, fever crises |
HSAN IV does NOT cause sensory ataxia — absent Romberg, intact proprioception. Excluded from this differential.
II. Hereditary Ataxias Where Sensory Neuropathy Is an Integral Feature (Presenting in Childhood)
1. Friedreich Ataxia (FA) — Most Common Hereditary Ataxia Worldwide
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | FXN (frataxin), chromosome 9q13 |
| Mutation | GAA trinucleotide repeat expansion in intron 1 (both alleles in most cases) |
| Onset | Childhood — first decade (mean ~15 years; occasionally 5–7 years) |
| Sensory component | Dorsal root ganglion neuronal degeneration → loss of vibration, proprioception, Romberg positive → sensory ataxia is the primary initial presentation |
| Cerebellar component | Also present (spinocerebellar tracts degenerate) — combined sensory + cerebellar ataxia |
| DTRs | Absent (areflexia — hallmark) |
| Plantar response | Extensor (Babinski sign) — co-existing pyramidal tract disease |
| Cardiomyopathy | Present in ~65% — major cause of death |
| Diabetes | ~25% |
| Deformities | Pes cavus, kyphoscoliosis |
| Pathogenesis | GAA repeat → frataxin ↓ → iron-sulfur cluster enzyme failure → mitochondrial oxidative stress |
FA is the paradigm of hereditary sensory ataxic neuropathy — the neuropathy (dorsal root ganglion degeneration) is what causes the sensory ataxia; cerebellar degeneration adds on later.
2. Ataxia with Vitamin E Deficiency (AVED)
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | TTPA (α-tocopherol transfer protein) |
| Onset | Childhood/adolescence — often mimics Friedreich ataxia precisely |
| Sensory features | Loss of proprioception and vibration → sensory ataxia; Romberg positive |
| Other features | Areflexia, Babinski sign, decreased visual acuity, head titubation |
| Key diagnostic test | Low serum vitamin E (α-tocopherol) with normal lipoproteins |
| Critical point | Treatable — vitamin E supplementation halts/reverses progression |
| Distinguish from FA | No cardiomyopathy; no GAA repeat; serum vitamin E low; normal frataxin |
3. Abetalipoproteinemia (Bassen-Kornzweig Syndrome)
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | MTTP (microsomal triglyceride transfer protein) |
| Onset | Infancy to childhood — can present very early |
| Primary defect | Absence of apolipoprotein B → no chylomicrons, VLDL → fat malabsorption |
| Secondary effect | Vitamin E (and A, D, K) deficiency → spinocerebellar and sensory neuropathy |
| Sensory features | Areflexia, proprioception loss, vibration loss → sensory ataxia |
| Other features | Acanthocytosis on blood smear, retinitis pigmentosa, steatorrhoea in infancy, coagulopathy |
| Key diagnostic test | Absent apolipoprotein B, acanthocytes on peripheral smear, extremely low cholesterol, triglycerides |
| Treatment | Fat-soluble vitamin supplementation (E, A, K, D) — halts neurological progression |
4. Ataxia-Telangiectasia (A-T)
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | ATM (chromosome 11q22–23) |
| Onset | Early childhood (~1–3 years) ✓ — one of the earliest-onset hereditary ataxias |
| Sensory component | Peripheral sensory neuropathy → contributes to sensory ataxia |
| Cerebellar component | Progressive cerebellar ataxia (primary feature early) |
| Other features | Oculomotor apraxia, choreoathetosis, myoclonus, dystonia, telangiectasias (conjunctiva/skin — appear at 3–5 years, after ataxia) |
| Immune deficiency | IgA/IgG deficiency → recurrent sinopulmonary infections |
| Cancer risk | T-cell leukemia/lymphoma (~30%) |
| Biomarker | Elevated AFP (alpha-fetoprotein) — key diagnostic clue |
| Radiation sensitivity | Contraindication to ionizing radiation |
5. Ataxia with Oculomotor Apraxia (AOA)
| Feature | Details |
|---|---|
| Inheritance | AR |
| Gene | APTX |
| Onset | Childhood (2–10 years) |
| Key features | Cerebellar ataxia + oculomotor apraxia + sensory-motor neuropathy → sensory ataxia |
| Biomarker | Low serum albumin, elevated cholesterol |
| Feature | Details |
|---|---|
| Inheritance | AR |
| Gene | SETX |
| Onset | Adolescence |
| Biomarker | Elevated AFP (distinguishes from AOA1) |
| Sensory neuropathy | Present → sensory ataxia component |
6. ARSACS (Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay)
| Feature | Details |
|---|---|
| Inheritance | AR |
| Gene | SACS (sacsin) |
| Onset | Early childhood — walking age (12–18 months) ✓ |
| Features | Spastic ataxia (combination of spasticity and cerebellar ataxia) + axonal sensorimotor polyneuropathy |
| Sensory ataxia | Proprioception loss contributing to ataxia |
| Retinal striations | Pathognomonic on fundoscopy |
| Founder population | Charlevoix-Saguenay region, Quebec (carrier frequency 1:22) |
7. SANDO / ANS (Sensory Ataxic Neuropathy, Dysarthria and Ophthalmoparesis / Ataxia-Neuropathy Spectrum — POLG-related)
| Feature | Details |
|---|---|
| Inheritance | AR (biallelic POLG1 mutations) |
| Gene | POLG1 (mitochondrial DNA polymerase) |
| Onset | Variable — can present in childhood (earlier onset with more severe mutations) |
| Sensory features | Sensory ataxic neuropathy — the primary and defining feature: large-fibre sensory loss → Romberg positive, absent proprioception |
| Other features | Dysarthria, external ophthalmoparesis (ptosis, ophthalmoplegia), epilepsy (especially SCAE spectrum), myopathy |
| Key warning | Valproate contraindicated — risk of fatal hepatotoxicity |
| Mitochondrial basis | Impaired mtDNA replication → ragged red fibres, COX-deficient fibres |
8. CANVAS (Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome)
| Feature | Details |
|---|---|
| Inheritance | AR |
| Gene | RFC1 (biallelic AAGGG pentanucleotide intronic expansion) |
| Onset | Usually middle age — not congenital |
| The triad | Cerebellar ataxia + sensory polyneuropathy (large fibres) + bilateral vestibular areflexia |
| Sensory ataxia | Core feature due to combined neuropathy + vestibular loss |
CANVAS typically does NOT present from birth — included here for completeness as a differential in adult-onset sensory ataxia.
9. Refsum Disease (Heredopathia Atactica Polyneuritiformis)
| Feature | Details |
|---|---|
| Inheritance | AR |
| Gene | PHYH (phytanoyl-CoA hydroxylase) or PEX7 |
| Onset | Childhood to early adulthood |
| Pathogenesis | Phytanic acid accumulation (from dairy, ruminant meat) → peripheral nerve and cerebellar damage |
| Sensory neuropathy | Large fibre polyneuropathy → sensory ataxia, areflexia |
| Other features | Retinitis pigmentosa (night blindness), anosmia, ichthyosis, sensorineural deafness, cardiomyopathy |
| Key point | Treatable — phytanic acid-restricted diet (avoid dairy, ruminant meat) + plasmapheresis in acute exacerbations |
| Diagnosis | Elevated plasma phytanic acid |
III. CMT with Sensory Ataxia Features
CMT Type 1A / 2 (Selected Subtypes)
- CMT2A (MFN2): Axonal; can have prominent sensory involvement
- CMT4 (recessive demyelinating): Earlier onset, more severe sensory involvement
- CMTX1 (GJB1, connexin 32): X-linked; sensory loss can be significant
CMT causes foot deformity and motor weakness prominently — purely sensory ataxic presentation is not the usual picture.
IV. Summary Table
| Condition | Inheritance | Gene | Onset | Sensory Ataxia Mechanism | Key Distinguishing Feature |
|---|---|---|---|---|---|
| Friedreich Ataxia | AR | FXN | 5–25 yrs | DRG neuronal degeneration → large fibre loss | Cardiomyopathy, diabetes, pes cavus, areflexia + Babinski |
| HSAN II | AR | WNK1/HSN2, FAM134B | Birth | Global sensory fibre loss incl. large fibres | Self-mutilation, absent all modalities |
| HSAN III (Riley-Day) | AR | IKBKAP | Congenital | Large fibre loss (later) | Ashkenazi, absent lacrimation, dysautonomia, no fungiform papillae |
| AVED | AR | TTPA | Childhood | Spinocerebellar degeneration (vitamin E deficiency) | Mimics FA; treatable; low serum vit E |
| Abetalipoproteinemia | AR | MTTP | Infancy | Fat malabsorption → vit E deficiency → DRG/dorsal column damage | Acanthocytes, steatorrhoea, low cholesterol, retinitis pigmentosa |
| Ataxia-Telangiectasia | AR | ATM | 1–3 yrs | Peripheral sensory neuropathy + cerebellar | Telangiectasia, ↑AFP, immune deficiency, cancer risk |
| AOA1 | AR | APTX | 2–10 yrs | Sensory-motor neuropathy | Low albumin, ↑cholesterol, oculomotor apraxia |
| AOA2 | AR | SETX | Adolescence | Sensory neuropathy | ↑AFP, oculomotor apraxia |
| ARSACS | AR | SACS | 12–18 months | Axonal sensorimotor neuropathy | Spastic ataxia, retinal striations, Quebec founder |
| SANDO/ANS (POLG) | AR | POLG1 | Childhood/adult | Sensory ataxic neuropathy (defining feature) | Ophthalmoparesis, epilepsy; valproate contraindicated |
| Refsum disease | AR | PHYH | Childhood | Peripheral neuropathy (large fibre) from phytanic acid | Retinitis pigmentosa, anosmia, ichthyosis; treatable |
| CANVAS | AR | RFC1 | Middle age | Sensory polyneuropathy + vestibular areflexia | Bilateral vestibular loss; dry cough |
V. Diagnostic Approach
Hereditary Sensory Ataxia from Birth / Early Life
│
├── Are ALL sensory modalities lost (large + small)?
│ └── HSAN II, HSAN III (congenital)
│ → Check: autonomic features, ethnicity, fungiform papillae
│
├── Large fibre predominantly (vibration, proprioception) + areflexia + normal pain?
│ └── FA, AVED, Abetalipoproteinemia, SANDO
│ → Check: cardiomyopathy, serum vitamin E, acanthocytes, POLG
│
├── Cerebellar + sensory + telangiectasia?
│ └── Ataxia-Telangiectasia → AFP, immunoglobulins
│
├── Oculomotor apraxia?
│ └── AOA1 (low albumin) or AOA2 (↑AFP)
│
├── Spastic + ataxic + very early?
│ └── ARSACS → fundoscopy (retinal striations)
│
├── Retinitis pigmentosa + anosmia + ichthyosis?
│ └── Refsum → plasma phytanic acid
│
└── Ophthalmoplegia + myopathy + epilepsy?
└── POLG/SANDO → muscle biopsy; avoid valproate
VI. Key Clinical Pearls
- Romberg positive + absent ankle jerks + extensor plantars in a child = Friedreich ataxia until proven otherwise
- AVED and abetalipoproteinemia are treatable — vitamin E supplementation can halt or reverse neurological decline; never miss these
- HSAN III (Familial Dysautonomia) is the only truly congenital-from-birth hereditary neuropathy with sensory ataxia — suspect in Ashkenazi infants with hypotonia, absent tear production, and dysautonomia
- AFP elevation is the key diagnostic marker distinguishing A-T from AOA2 from other cerebellar ataxias
- POLG/SANDO is the mitochondrial differential for sensory ataxic neuropathy — always check before prescribing valproate in any ataxia-epilepsy patient
- Refsum disease is completely preventable with dietary restriction once diagnosed — never miss it in ataxia + retinitis pigmentosa + ichthyosis
Hemorrhagic transformation in a ischemic stroke grades
hemorrhagic transformation ischemic stroke ECASS classification HI1 HI2 PH1 PH2 grading criteria CT MRI
hemorrhagic transformation grading ECASS classification HI1 HI2 PH1 PH2 risk factors management symptomatic
Heidelberg bleeding classification hemorrhagic transformation stroke 2015 categories management sICH definition
hemorrhagic transformation risk factors management anticoagulation restart antithrombotic after sICH
Hemorrhagic Transformation (HT) in Ischemic Stroke — Classification and Grading
Definition and Pathophysiology
- Ischemia damages endothelial tight junctions and the blood-brain barrier (BBB)
- Reperfusion (spontaneous or therapeutic) restores blood flow to vessels whose integrity is already compromised
- Blood extravasates — ranging from petechiae to frank hematoma
- Thrombolytics (tPA) and mechanical thrombectomy accelerate and amplify this process
"Secondary hemorrhage can occur from ischemia-reperfusion injury following spontaneous or therapeutic dissolution or fragmentation of the intravascular occlusive material... resulting reperfusion hemorrhages are largely punctate but may be multiple or even confluent." — Robbins & Cotran Pathologic Basis of Disease
The Two Major Classification Systems
1. ECASS Classification (European Cooperative Acute Stroke Study)
Category A: Hemorrhagic Infarction (HI) — No/Minimal Mass Effect
HI-1: Hemorrhagic Infarction Type 1
| Feature | Description |
|---|---|
| Appearance | Small, scattered petechiae along the margins of the infarcted area |
| Distribution | Peripheral; at the edge of the infarct zone |
| Mass effect | None |
| Volume | Minimal |
| Clinical significance | Almost always asymptomatic |
| Management | No specific intervention required |
| Pathology | Superficial cortical reperfusion into the border zone |
HI-2: Hemorrhagic Infarction Type 2
| Feature | Description |
|---|---|
| Appearance | Confluent petechiae throughout the infarcted area (not just margins) |
| Distribution | Throughout the infarct, merging together |
| Mass effect | None |
| Volume | More extensive than HI-1, still petechial |
| Clinical significance | Usually asymptomatic; neurological worsening usually from edema/infarct progression, not the bleed itself |
| Management | No specific intervention; does not count as sICH in most trial definitions |
Category B: Parenchymal Hemorrhage (PH) — WITH Mass Effect
PH-1: Parenchymal Hemorrhage Type 1
| Feature | Description |
|---|---|
| Appearance | Hyperdense hematoma within the infarcted tissue |
| Volume | Occupies ≤30% of the infarcted area |
| Mass effect | Mild (slight displacement of structures) |
| Clinical significance | May or may not be symptomatic; associated with some worsening |
| Management | Close monitoring; hold antithrombotics; individualized decision on BP management |
PH-2: Parenchymal Hemorrhage Type 2
| Feature | Description |
|---|---|
| Appearance | Hyperdense hematoma — large, dense blood clot |
| Volume | Occupies >30% of the infarcted area |
| Mass effect | Definite/marked — midline shift, herniation risk |
| Clinical significance | Almost always symptomatic; this is the definition of sICH (symptomatic intracranial hemorrhage) in most trials |
| Management | Emergency: BP control, reverse anticoagulation, neurosurgical consultation; high mortality |
| Prognosis | Mortality ~50%; significant neurological deterioration in survivors |
ECASS Summary Table
| Type | Petechial/Hematoma | Location | Mass Effect | Symptomatic | Volume |
|---|---|---|---|---|---|
| HI-1 | Petechial (scattered) | Margins of infarct | None | Rarely | Minimal |
| HI-2 | Petechial (confluent) | Throughout infarct | None | Rarely | Moderate |
| PH-1 | Hematoma | Within infarct | Mild | Sometimes | <30% of infarct |
| PH-2 | Hematoma | Within infarct | Marked/definite | Almost always | >30% of infarct |
2. Heidelberg Bleeding Classification (2015, von Kummer et al.)
| Class | Type | Criteria |
|---|---|---|
| 1a | HI-1 | Scattered small petechiae, no mass effect |
| 1b | HI-2 | Confluent petechiae, no mass effect |
| 1c | PH-1 | Hematoma within infarcted tissue, <30% of infarct, no substantive mass effect |
| 2 | PH-2 | Hematoma occupying ≥30% of infarcted tissue, obvious mass effect |
| 3a | Remote PH | Parenchymal hematoma remote from infarcted brain tissue |
| 3b | IVH | Intraventricular hemorrhage |
| 3c | SAH | Subarachnoid hemorrhage |
| 3d | SDH | Subdural hemorrhage |
Classes 3a–3d represent bleeds outside the infarct territory — typically iatrogenic complications of catheter-based thrombectomy or systemic thrombolysis.
Pathological Appearance
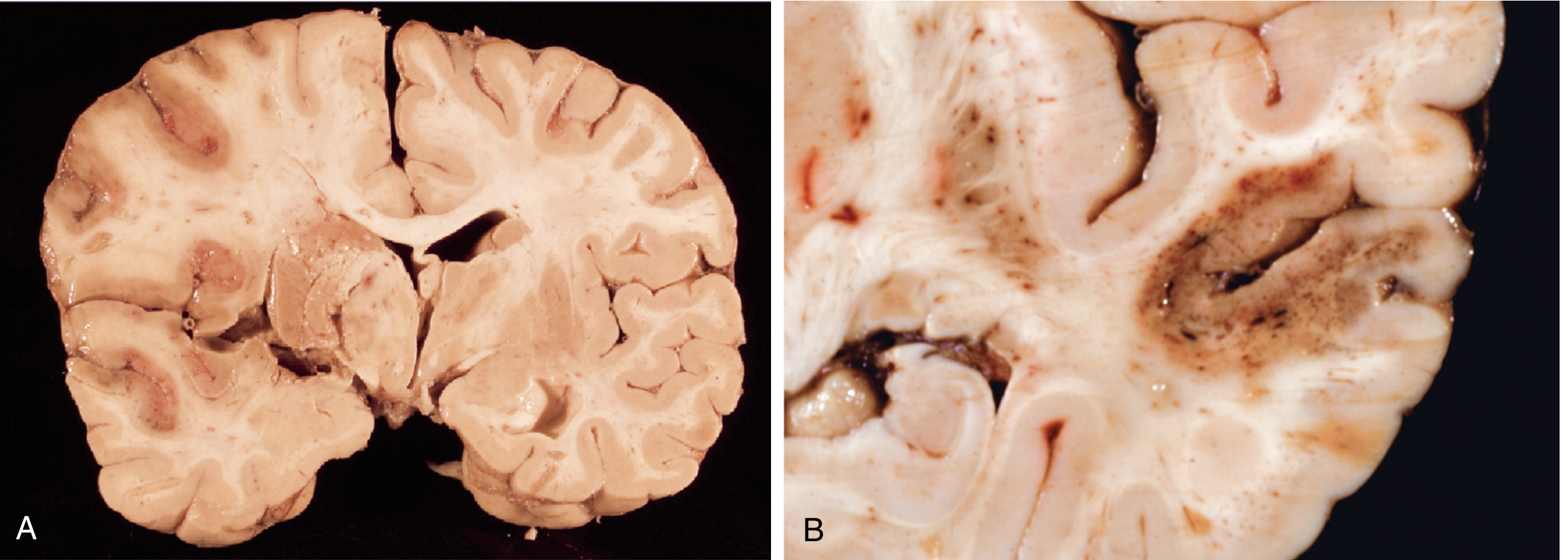
Definitions of Symptomatic ICH (sICH) — Trial-Specific
| Trial | Definition of sICH |
|---|---|
| ECASS III | Any ICH + NIHSS increase ≥4 points, where ICH is judged to have caused deterioration |
| NINDS | Any hemorrhage on CT ± any neurological worsening |
| SITS-MOST | ICH type PH-2 + NIHSS increase ≥4 points within 24 h |
| PROACT II | Any hemorrhage + NIHSS increase ≥4 points |
⚠️ Only PH-2 is consistently linked to neurological worsening and death. HI-1 and HI-2 are largely imaging findings and are usually clinically silent.
Timing of HT
| Setting | Typical Timing |
|---|---|
| Spontaneous (no reperfusion therapy) | First 4 days; rarely in first 6 hours |
| After IV thrombolysis (tPA) | Usually within 24 hours of treatment |
| After mechanical thrombectomy | Within 24–36 hours |
| MRI detects HT earlier than CT (GRE/SWI sequences) | — |
Risk Factors for HT
Patient/Infarct Factors
| Factor | Direction |
|---|---|
| Large infarct volume (DWI) | ↑ Risk |
| Cardioembolic stroke (especially AF) | ↑ Risk (highest spontaneous HT risk) |
| Early ischemic changes on baseline CT | ↑ Risk |
| Hyperglycemia | ↑ Risk |
| Hypertension (uncontrolled) | ↑ Risk |
| Age >80 | ↑ Risk |
| Leukoaraiosis (white matter disease) | ↑ Risk |
| Cerebral microbleeds on MRI | ↑ Risk |
| BBB permeability (HARM on MRI) | ↑ Risk |
| Low collateral flow | ↑ Risk |
| High NIHSS at presentation | ↑ Risk |
Treatment Factors
| Factor | Direction |
|---|---|
| IV thrombolysis (tPA) | ↑ Risk (6–8% sICH) |
| Mechanical thrombectomy (multiple passes) | ↑ Risk |
| Anticoagulation use | ↑ Risk |
| Combined IV + IA therapy | ↑ Risk |
Management by Grade
| Grade | Immediate Action |
|---|---|
| HI-1 | Monitor clinically; no specific intervention; continue antiplatelet with caution |
| HI-2 | Monitor; hold anticoagulants temporarily; reassess imaging in 24–48 h |
| PH-1 | Hold anticoagulants/antithrombotics; BP control (<180/105 mmHg); neurology consultation; repeat imaging |
| PH-2 | Emergency management: reverse any coagulopathy (PCC, FFP, protamine if heparin, idarucizumab if dabigatran); aggressive BP control; neurosurgical consultation for decompression if mass effect threatening herniation; ICU admission |
Antithrombotic Restart After sICH
- Antiplatelet (aspirin): restart after 1–4 weeks in most cardioembolic/HI-1/HI-2 cases
- Anticoagulation (for AF): restart after 2–4 weeks if PH-2 has stabilised — weigh stroke recurrence risk vs. rebleed risk
- Never restart anticoagulation if remote PH (class 3a) or large PH-2 without dedicated multidisciplinary review
Summary: The 4 ECASS Grades at a Glance
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━
HEMORRHAGIC INFARCTION (No mass effect — petechial)
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━
HI-1: Scattered petechiae at MARGINS of infarct
→ Asymptomatic | No treatment needed
HI-2: Confluent petechiae THROUGHOUT infarct
→ Usually asymptomatic | Monitor only
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━
PARENCHYMAL HEMATOMA (WITH mass effect — blood clot)
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━
PH-1: Hematoma <30% of infarct | Mild mass effect
→ Sometimes symptomatic | Hold antithrombotics, monitor
PH-2: Hematoma >30% of infarct | Marked mass effect
→ Almost always symptomatic (= sICH)
→ EMERGENCY: reverse coagulopathy, BP control, neuro Sx
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━
Treatment according to grades