explain mechanism of cerebral auto regulation
Reading File
Finding Sources
Finding Sources
Searching PubMed
cerebral autoregulation mechanism
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have all the information needed for a comprehensive, well-illustrated answer. Here it is:
Cerebral Autoregulation — Mechanism
Definition & Physiological Range
Cerebral autoregulation (CA) is the intrinsic ability of the cerebral vasculature to maintain a relatively constant cerebral blood flow (CBF) (~50 mL/100 g/min) across a wide range of cerebral perfusion pressures (CPP). In normal individuals, this plateau is maintained between CPPs of approximately 50–150 mmHg (or MAPs of ~60–160 mmHg). Outside these limits, CBF becomes pressure-passive — falling with hypotension and rising with hypertension.
CPP = MAP − ICP
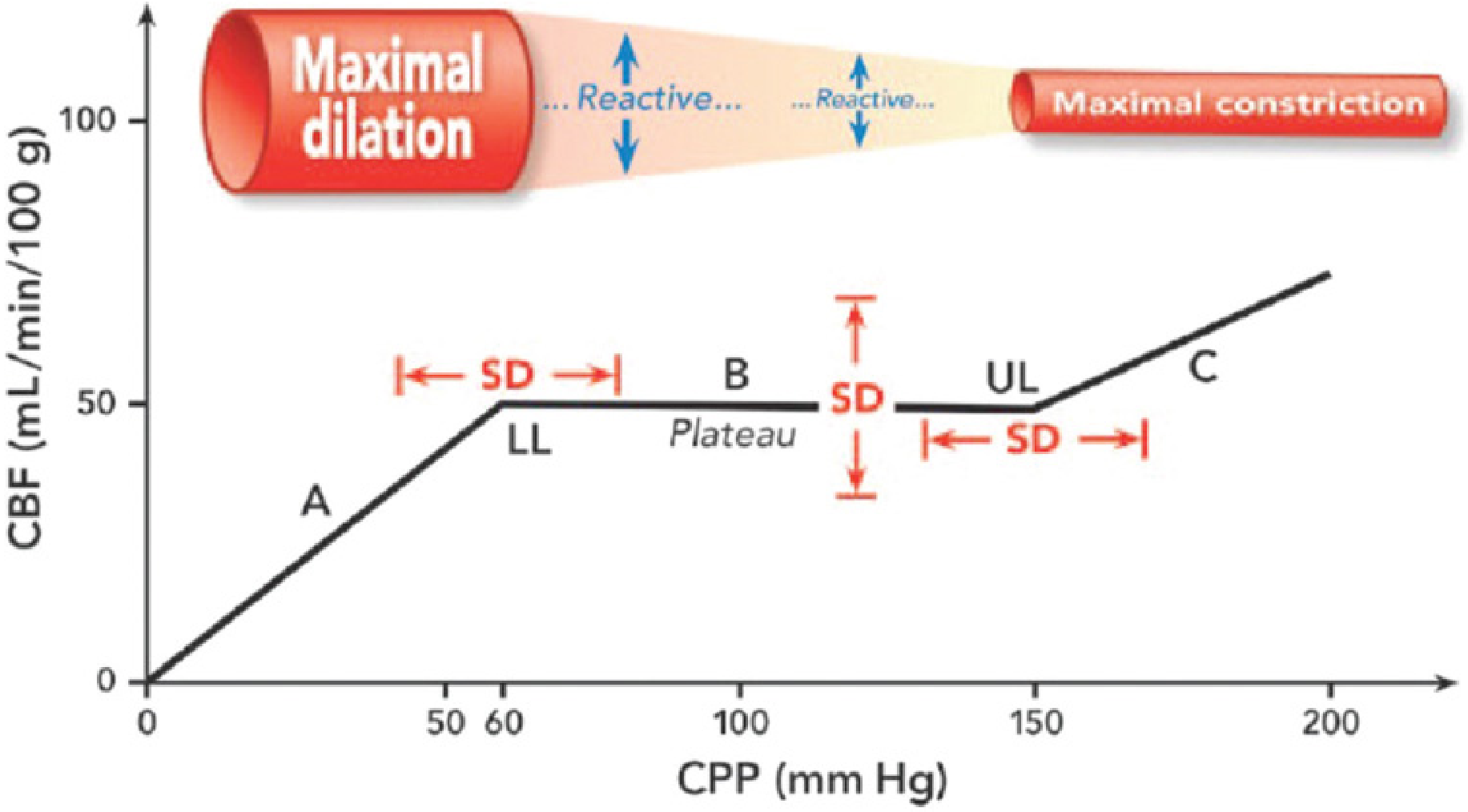
Three Core Mechanisms
1. Myogenic Mechanism
The myogenic response is the most fundamental component. Cerebral vascular smooth muscle cells intrinsically sense wall stretch:
- ↑ CPP → vessel wall stretch → smooth muscle depolarization → Ca²⁺ influx → vasoconstriction
- ↓ CPP → reduced wall tension → smooth muscle relaxation → vasodilation
This response is rapid (10–60 seconds) and does not require neural input. It operates primarily at the level of small arteries and arterioles. The Bayliss effect (intrinsic vascular smooth muscle contraction in response to stretch) is the cellular basis.
2. Metabolic / Flow-Metabolism Coupling Mechanism
Regional CBF is tightly coupled to local metabolic demand — a process regulated within the neurovascular unit (NVU), which comprises neurons, astrocytes, microglia, pericytes, endothelial cells, and the basement membrane.
Key vasoactive mediators generated with increased neuronal activity:
| Mediator | Source | Vascular Effect |
|---|---|---|
| Nitric oxide (NO) | Neuronal NOS (nNOS) via NMDA-R/Ca²⁺ activation; endothelial eNOS | Vasodilation via cGMP |
| Prostaglandins (PGE₂) | Neuronal PLA₂ → AA → COX pathway | Vasodilation |
| EETs (epoxyeicosatrienoic acids) | Astrocyte mGluR → Ca²⁺ → PLA₂ → AA | Vasodilation |
| Adenosine | ATP breakdown in energy-deficient states | Vasodilation |
| K⁺, H⁺, Lactate | Local metabolic by-products | Vasodilation |
| 20-HETE | AA via cytochrome P450 | Vasoconstriction (counterbalances) |
Astrocytes are pivotal: glutamate activates metabotropic glutamate receptors (mGluR) on astrocyte endfeet → Ca²⁺ waves → release of EETs and PGE₂ → smooth muscle dilation. The vasodilatory signal then propagates retrogradely via gap junctions along the endothelium and smooth muscle of penetrating arterioles back to pial arteries, reducing proximal resistance and increasing regional flow precisely where neural activity is greatest.
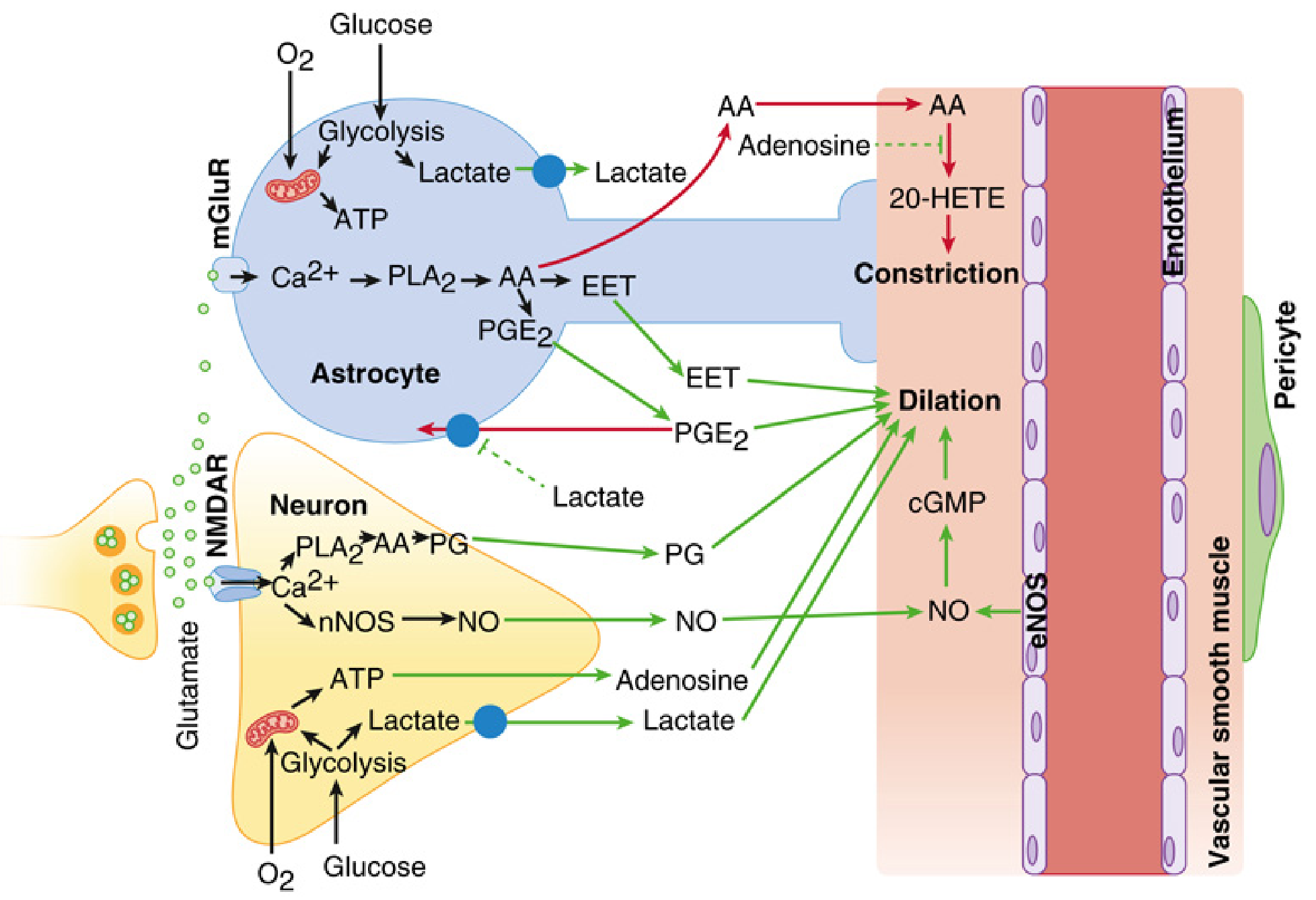
3. Neurogenic Mechanism
Cerebral vessels receive perivascular innervation from:
- Sympathetic fibers (from superior cervical ganglion): release norepinephrine, neuropeptide Y → vasoconstriction
- Parasympathetic fibers (from sphenopalatine/otic ganglia): release ACh, VIP → vasodilation
- Sensory fibers (from trigeminal ganglion): release substance P, CGRP → vasodilation
The sympathetic nervous system shifts the autoregulation curve rightward during hypertension, providing cerebrovascular protection at high pressures. However, it simultaneously reduces vasodilatory capacity during hypotension. Perivascular neurons also release vasoactive intestinal peptide (VIP), somatostatin, cholecystokinin, and calcitonin gene-related peptide, which modulate tone locally.
Modifying Factors
| Factor | Effect on CA |
|---|---|
| PaCO₂ | Most potent extrinsic factor. CBF ∝ PaCO₂ between 20–80 mmHg (~1–2 mL/100g/min per mmHg). CO₂ diffuses freely across BBB → alters CSF/tissue pH → vasomotor response |
| PaO₂ | Hypoxia (PaO₂ <50 mmHg) markedly increases CBF; hyperoxia causes minor decrease |
| Chronic hypertension | Shifts curve rightward — both lower and upper limits increase; flow becomes pressure-dependent at "normal" MAPs |
| TBI / CNS injury | Disrupts CA in ~1/3 of severe TBI patients — CBF becomes pressure-passive |
| Volatile anesthetics | Impair CA dose-dependently; cerebral vasculature retains CO₂ reactivity |
| ICP elevation | Cushing reflex: brainstem ischemia → sympathetic surge → ↑MAP to maintain CPP |
Contemporary (Dynamic) View
The classical view of a static plateau is now considered outdated. CA is a dynamic process reflecting the integrated balance of:
- Myogenic autoregulation
- Neurovascular coupling
- Arterial CO₂/O₂ tensions
- Autonomic neurogenic tone
- Cardiac output (increasingly recognized as a CPP determinant)
Static autoregulation (measured over ~10 min) shows a wide plateau; dynamic autoregulation (measured over seconds with transient BP changes) shows a much narrower plateau (as little as 10 mmHg) because buffering is time-dependent. There is also substantial inter-individual variation in both limits and the width of the plateau.
Clinical Correlates
- Below LLA (~CPP <50–60 mmHg): maximal vasodilation is exhausted → ischemia. Symptoms appear when CBF falls 35–40% below baseline.
- Above upper limit (~CPP >150 mmHg): forced dilation of arterioles → disruption of blood–brain barrier → cerebral edema, hemorrhage (hypertensive encephalopathy).
- TBI management: Target CPP 60–70 mmHg; higher CPP targets increase ARDS risk without neurologic benefit.
- Pressure reactivity index (PRx): continuous correlation between ICP and MAP waves — used to monitor real-time autoregulatory status in ICU patients.
Sources: Miller's Anesthesia, 10e (Ch. 10); Morgan & Mikhail's Clinical Anesthesiology, 7e (Ch. 26); Medical Physiology – Boron & Boulpaep (Ch. 24); Mulholland and Greenfield's Surgery, 7e (Ch. 21)
explain metabolic mechanism again in simple words
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.