I now have comprehensive, textbook-quality content from Robbins & Cotran, Thompson & Thompson Genetics, and Emery's Genetics. Let me compose the detailed notes now.
DOWN SYNDROME - Detailed Exam Notes
Part 1 of Many: Introduction, Etiology & Pathogenesis
SUBTOPIC 1: INTRODUCTION & EPIDEMIOLOGY
What is Down Syndrome?
Down syndrome (DS) is the single most common chromosomal disorder in humans and the most common genetic cause of moderate intellectual disability. It was first described clinically by John Langdon Down in 1866 and later linked to trisomy of chromosome 21 by Jerome Lejeune in 1959.
Key identifiers for MCQs:
- Also called: Trisomy 21 (most common form), Down syndrome, mongolism (obsolete term - never use clinically)
- Chromosome affected: Chromosome 21 (smallest autosome; gene-poor, high AT:GC ratio)
- Most common chromosomal cause of intellectual disability
- Most common autosomal trisomy compatible with survival to adulthood
Incidence & Epidemiology
| Parameter | Value |
|---|
| Overall incidence (live births) | ~1 in 700 |
| Risk at maternal age < 20 years | 1 in 1550 |
| Risk at maternal age 30 years | begins to rise sharply |
| Risk at maternal age 35 years | ~1 in 350 |
| Risk at maternal age > 45 years | ~1 in 25 |
| Survival to age 20 | ~90% of live-born individuals |
| Median age at death (modern era) | 47 years (up from 25 years in 1983) |
| Average life expectancy (without severe cardiac anomaly) | 50-60 years |
Why Does Incidence Rise With Maternal Age? (Examiner Favorite)
The strong correlation with maternal age is one of the most tested concepts. Here is the full reasoning:
- Oocytes are arrested in prophase I of meiosis from fetal life onwards - they resume meiosis only at ovulation, decades later
- This prolonged meiotic arrest makes the oocyte vulnerable to cohesin degradation over time - cohesins are the proteins that hold homologous chromosomes together; as a woman ages, cohesin levels decline, increasing the chance of premature separation
- This leads to meiotic nondisjunction of chromosome 21
- ~95% of extra chromosome 21s are of maternal origin (confirmed by DNA polymorphism studies)
- Of maternal errors, ~70-80% occur in Meiosis I (the first division), which is the most vulnerable step
- Only ~10% of nondisjunction events are of paternal origin
Exam Trap: Even though older mothers have MUCH higher risk, they contribute a MINORITY of DS births in the population - because younger mothers have far higher birth rates. More than half of all DS babies are born to mothers younger than 35. This is why screening programs target all pregnant women, not just older ones.
Comparative Risk - Autosomal Trisomies (High-Yield Table)
| Feature | Trisomy 21 (Down) | Trisomy 18 (Edwards) | Trisomy 13 (Patau) |
|---|
| Incidence | 1 in 700 | 1 in 6,000-8,000 | 1 in 5,000-15,000 |
| Chromosome | 21 | 18 | 13 |
| Major feature | Intellectual disability, flat face | Clenched fists, rocker-bottom feet | Holoprosencephaly, cleft lip/palate, polydactyly |
| Cardiac defects | ~40-50% | ~90% | ~90% |
| Survival | Most reach adulthood | Days to weeks (rarely months) | Days to weeks |
| Maternal age effect | Yes | Yes | Yes |
SUBTOPIC 2: ETIOLOGY - THE THREE KARYOTYPIC TYPES
This is the most heavily tested section in genetics examinations. You must know all three types, their percentages, karyotypes, mechanisms, maternal age correlation, and recurrence risks cold.
Type 1: Free Trisomy 21 (~95% of all cases)
Karyotype: 47,XX,+21 or 47,XY,+21
Mechanism: Meiotic nondisjunction of chromosome 21
During normal meiosis, homologous chromosomes (in meiosis I) or sister chromatids (in meiosis II) separate properly to opposite poles. Nondisjunction is the failure of this separation, resulting in one daughter cell receiving both copies and the other receiving none.
Where does nondisjunction occur?
- Maternal Meiosis I: ~70-80% of cases (most common error)
- Maternal Meiosis II: ~20-25% of cases
- Paternal Meiosis (either division): ~10% of cases
Result of fertilization:
- Trisomic egg + normal sperm = Trisomy 21 conceptus (47 chromosomes, 3 copies of chromosome 21)
- Nullisomic egg + normal sperm = Monosomy 21 (not viable, spontaneously aborted)
Key points for exams:
- Parents have completely normal karyotypes (46 chromosomes each)
- Strongly correlated with advanced maternal age
- Recurrence risk: ~1% above the age-related risk (i.e., about 1 in 100 additional risk beyond baseline)
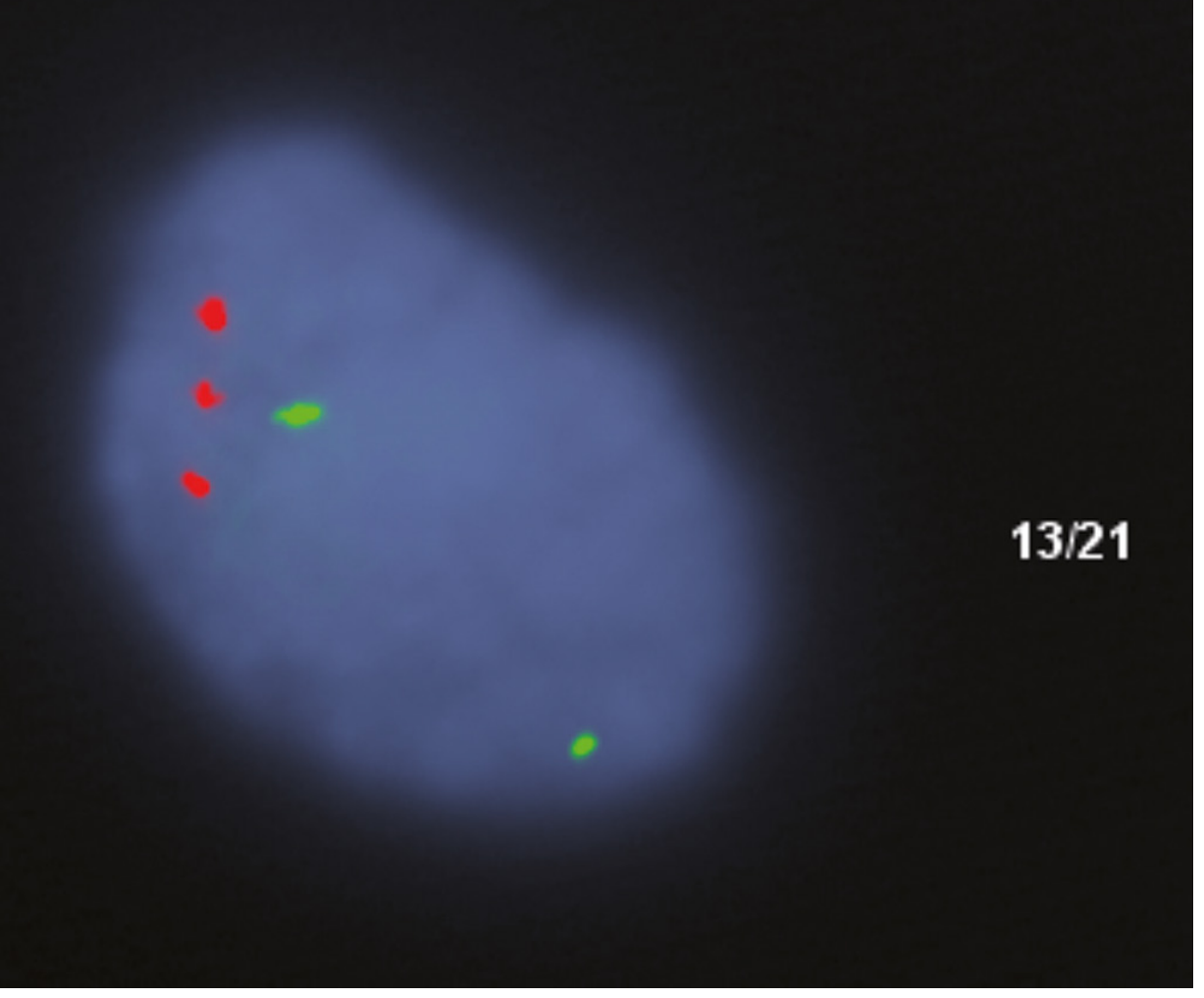
- FISH with chromosome 21-specific probes shows 3 red signals in interphase nuclei (see image below)
FISH analysis using locus-specific probes showing 3 red signals (chromosome 21) confirming trisomy 21 - Robbins & Cotran Pathologic Basis of Disease
Type 2: Robertsonian Translocation (~4% of all cases)
Karyotype: 46 chromosomes (not 47!) - e.g., 46,XX,rob(14;21)(q10;q10),+21
This is the MOST IMPORTANT TYPE FOR MCQs and vivas because:
- The chromosome count is 46, not 47 - a classic trap
- It has a familial/hereditary pattern
- It has the highest recurrence risk
- It is NOT related to maternal age
Mechanism:
- Robertsonian translocation = fusion of the long arms (q arms) of two acrocentric chromosomes at their centromeres, with loss of the (genetically insignificant) short arms
- Acrocentric chromosomes: 13, 14, 15, 21, 22
- Most common partner for chromosome 21 in DS: Chromosome 14 (less commonly chromosome 22)
- The resulting translocation chromosome carries the genetic material of both 21q and 14q
In the affected Down syndrome patient:
- They have 46 chromosomes but are functionally trisomic for chromosome 21q
- They have: 2 normal chromosome 21s + 1 translocation chromosome (which contains another copy of 21q fused to 14q)
- Net result = triple gene dosage for chromosome 21 genes - same as free trisomy 21 in terms of phenotype
Carrier parent (Balanced Robertsonian carrier):
- Has only 45 chromosomes
- Karyotype: 45,XX,rob(14;21)(q10;q10) - they are phenotypically NORMAL
- Usually the mother is the carrier (more commonly maternal transmission)
- The carrier has only ONE chromosome 14 and ONE chromosome 21, replaced by the single translocation chromosome
Gametes produced by a carrier parent (Figure 6.3 from Thompson & Thompson - must know this):
Theoretically 6 gamete types, but only 3 lead to viable offspring:
- Normal gamete - normal child
- Balanced carrier gamete - phenotypically normal carrier child (like the parent)
- Unbalanced gamete (extra chromosome 21 material) - Down syndrome child
Theoretical recurrence risk = 1 in 3 for DS offspring. However, empirical population data shows much lower rates due to selection against unbalanced gametes and embryos:
- Carrier MOTHER: ~10-15% risk for DS offspring
- Carrier FATHER: ~2-5% risk for DS offspring (lower due to selection against unbalanced sperm)
Why is this critical in genetic counseling?
- When DS is diagnosed in a child with translocation karyotype, BOTH PARENTS MUST BE KARYOTYPED
- If a parent is a balanced carrier, siblings and other relatives should also be offered karyotyping
- This type has NO relation to maternal age
Type 3: Mosaic Down Syndrome (~1-2% of cases)
Karyotype: Mixture - e.g., 47,XX,+21/46,XX (two cell populations)
Mechanism: Mitotic nondisjunction during early embryogenesis AFTER fertilization
- A normal 46-chromosome zygote undergoes a faulty mitotic division
- One daughter cell gets 3 copies of chromosome 21 (47) and the other gets 1 copy (45)
- The 45-chromosome cell line (monosomy 21) is not viable and is eliminated
- The 47-chromosome line persists alongside the normal 46-chromosome line
- Result: mosaic individual with a mix of normal and trisomic cells
Clinical significance:
- Phenotype is generally MILDER than full trisomy 21
- Severity depends on proportion of trisomic cells and the tissues/organs affected
- Some mosaic individuals have near-normal intelligence
- IQ scores tend to be higher than in full trisomy 21 (range 40-75 in full trisomy vs. can be near-normal in mosaics)
- Maternal age is NOT a significant factor (this is a post-fertilization event)
Summary Table: Three Karyotypic Types (Exam Gold)
| Feature | Free Trisomy 21 | Robertsonian Translocation | Mosaic |
|---|
| Frequency | ~95% | ~4% | ~1% |
| Chromosome count | 47 | 46 | 46/47 mix |
| Mechanism | Meiotic nondisjunction | Unbalanced segregation of parental translocation | Mitotic nondisjunction post-fertilization |
| Maternal age effect | YES | NO | NO |
| Familial? | NO (sporadic) | Often YES (check parents) | NO |
| Recurrence risk | ~1% + maternal age risk | 10-15% (if mother is carrier); 2-5% (father is carrier) | Low |
| Parent karyotype | Both normal (46) | Parent may be balanced carrier (45 chromosomes) | Both normal (46) |
| Phenotype | Full DS | Full DS (clinically identical) | Variable, often milder |
Special Sub-types Worth Knowing
21q21q Translocation (isochromosome 21q):
- Rare, but critically important for counseling
- A carrier parent produces gametes that MUST EITHER contain a double dose of 21q OR no chromosome 21 at all
- Offspring are either Down syndrome or monosomy 21 (lethal)
- Recurrence risk = 100% - no normal children possible from a carrier of this chromosome
- Often arises de novo but when familial, requires urgent counseling
Partial Trisomy 21:
- Only a portion of 21q is present in triplicate
- Used in research to map genotype-phenotype correlations
- A region at the distal end of 21q (21q22) appears critical for the typical facial features
- A region less than 2 Mb has been identified as critical for the congenital heart defects seen in ~40% of DS patients
SUBTOPIC 3: PATHOGENESIS - HOW DOES AN EXTRA CHROMOSOME 21 CAUSE ALL THE FEATURES?
This is where conceptual understanding separates you from your peers who only memorize features. This is also increasingly tested in modern exams.
The Core Principle: Gene Dosage Effects
Down syndrome is fundamentally a gene dosage disorder - it is not the presence or absence of any single gene, but the 50% overexpression of hundreds of genes on chromosome 21 simultaneously, affecting multiple developmental pathways in different tissues at different times.
Chromosome 21 contains approximately 200-300 protein-coding genes and has the highest density of long non-coding RNAs (lncRNAs) of any chromosome. The long arm was fully sequenced in 2000, but unraveling which genes cause which features remains an active area of research.
Key Molecular Mechanisms
1. Gene Dosage and Overexpression
Most protein-coding genes on chromosome 21 are overexpressed by ~1.5-fold (50% more than normal). This seemingly small increase has profound developmental consequences because:
- Critical developmental thresholds are disrupted
- Cell signaling pathways are dysregulated
- Protein-protein interaction networks are disturbed by stoichiometric imbalance
2. APP Gene and Alzheimer Disease
Gene: Amyloid Precursor Protein (APP) gene - located on chromosome 21
Mechanism:
- With 3 copies of chromosome 21, there are 3 copies of APP
- APP is the precursor to amyloid-beta (Aβ) peptides
- Overproduction of Aβ leads to its aggregation and deposition as amyloid plaques in the brain
- This is the same mechanism implicated in familial early-onset Alzheimer disease
- Result: virtually ALL individuals with DS over age 40 develop neuropathological changes of Alzheimer disease (senile plaques, neurofibrillary tangles, cortical atrophy, ventricular dilatation)
- Clinical dementia typically appears 1-2 decades earlier than in the general population
Examiner's Gem: People with DS who have only a partial trisomy 21 NOT including the APP locus do NOT develop early-onset Alzheimer disease - confirming that APP gene dosage is responsible, not the trisomy state per se.
3. Mitochondrial Dysfunction
Approximately 10% of genes overexpressed in DS are directly or indirectly involved in regulating mitochondrial function. Consequences:
- Mitochondria show broken or swollen cristae on electron microscopy
- Increased generation of reactive oxygen species (ROS)
- Activation of apoptotic pathways
- This contributes to intellectual disability, premature aging, and tissue damage
4. Leukemia Predisposition
- GATA1 gene on the X chromosome is a key regulator of megakaryocyte and erythroid differentiation
- In DS, the trisomic environment during fetal development promotes clonal expansion of abnormal megakaryocyte-erythroid progenitors
- Specifically, fetal DS cells with mutations in GATA1 undergo clonal expansion causing Transient Myeloproliferative Disorder (TMD) in newborns
- TMD is unique to DS and resolves spontaneously in most cases within 3 months
- However, ~20-30% of TMD cases progress to Acute Megakaryoblastic Leukemia (AMKL) within 1-3 years
- Leukemia risk overall in DS:
- 20-fold increased risk for precursor B-cell ALL
- 500-fold increased risk for Acute Myeloid Leukemia (specifically AMKL)
5. Cardiac Defect Pathogenesis
- The ~2 Mb critical region on 21q22 contains genes critical for cardiac morphogenesis
- DSCAM (Down Syndrome Cell Adhesion Molecule) and DYRK1A kinase are among the candidates
- These disrupt endocardial cushion development, explaining the high rate of atrioventricular septal defects
6. Noncoding RNAs
Chromosome 21 has the highest density of lncRNAs of any chromosome. Their functions remain largely unknown, but their overexpression is thought to contribute to the broad phenotypic effects through regulation of gene expression networks.
7. Down Syndrome Critical Region (DSCR)
- Located at distal 21q22
- Children with partial trisomy limited to this region show typical Down syndrome facial features
- However, the concept of a single "critical region" is now considered an oversimplification - the full phenotype requires trisomy of multiple regions
- DYRK1A (Dual-specificity Tyrosine-phosphorylation Regulated Kinase 1A) in this region:
- Involved in neuronal development
- Overexpression disrupts dendritic spine development and synaptic plasticity
- Key contributor to intellectual disability
8. Immune Dysregulation
- Mainly affects T-cell functions
- Predisposes to serious infections, especially pulmonary infections (leading cause of death after the neonatal period)
- Also predisposes to thyroid autoimmunity - Hashimoto's thyroiditis is significantly more common in DS
- The molecular basis is not fully understood but relates to chromosome 21 gene effects on immune cell development
Genotype-Phenotype Correlation Summary
| Feature | Gene(s) Implicated | Chromosome 21 Region |
|---|
| Alzheimer disease (early onset) | APP | 21q21 |
| Intellectual disability | DYRK1A, DSCAM, RCAN1 | 21q22 |
| Congenital heart disease | DSCAM, COL6A1 (possibly) | 21q22 |
| Acute Megakaryoblastic Leukemia | GATA1 (mutation in context of trisomy 21) | GATA1 is on X chromosome |
| Facial features | Multiple genes in DSCR | 21q22 |
How Gene Imbalance Drives the Phenotype - The Big Picture
From Thompson & Thompson Genetics (p. 103): "It is important to think of this constellation of clinical findings, their variation, and likely outcomes in terms of gene imbalance - the relative overabundance of specific gene products; their impact on various critical pathways in particular tissues and cell types, both early in development and throughout life; and the particular alleles present in an individual's genome."
This means:
- The same trisomy 21 karyotype can produce variable phenotypes because other genes in the rest of the genome (from both parents) modify the effects of chromosome 21 overexpression
- This explains why some DS individuals are more severely affected than others, even with the same karyotype
- It also explains why mosaics are variable - the proportion of trisomic cells AND which tissues carry them matters
Quick-Fire Exam Facts for This Section
- Down syndrome chromosome 21 was fully sequenced in 2000
- Chromosome 21 is gene-poor with a high AT:GC ratio
- Extra chromosome 21 is of maternal origin in 95% of cases
- Most common meiotic error is in Maternal Meiosis I
- Nondisjunction is linked to cohesin degradation in aging oocytes
- APP gene dosage explains early Alzheimer disease
- GATA1 mutation + trisomy 21 = Transient Myeloproliferative Disorder
- Chromosome 21 has the highest lncRNA density of any chromosome
- DSCR at 21q22 is critical for facial features and cardiac defects
Sources Used
- Robbins & Cotran Pathologic Basis of Disease (p. 163-165)
- Thompson & Thompson Genetics and Genomics in Medicine, 9th edition (p. 102-104)
- Emery's Elements of Medical Genetics and Genomics (p. 263)
That covers the first three subtopics thoroughly. Whenever you are ready, say "next" and I will move on to the next three subtopics:
Coming Up Next:
- Subtopic 4: Clinical Features - Head to Toe (The Full Phenotype)
- Subtopic 5: Associated Conditions and Systemic Complications
- Subtopic 6: Diagnosis - Prenatal Screening and Postnatal Confirmation