Urea cycle
Reading File
Finding Sources
Searching the Web
urea cycle biochemistry steps enzymes overview
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content. Let me compile a thorough answer.
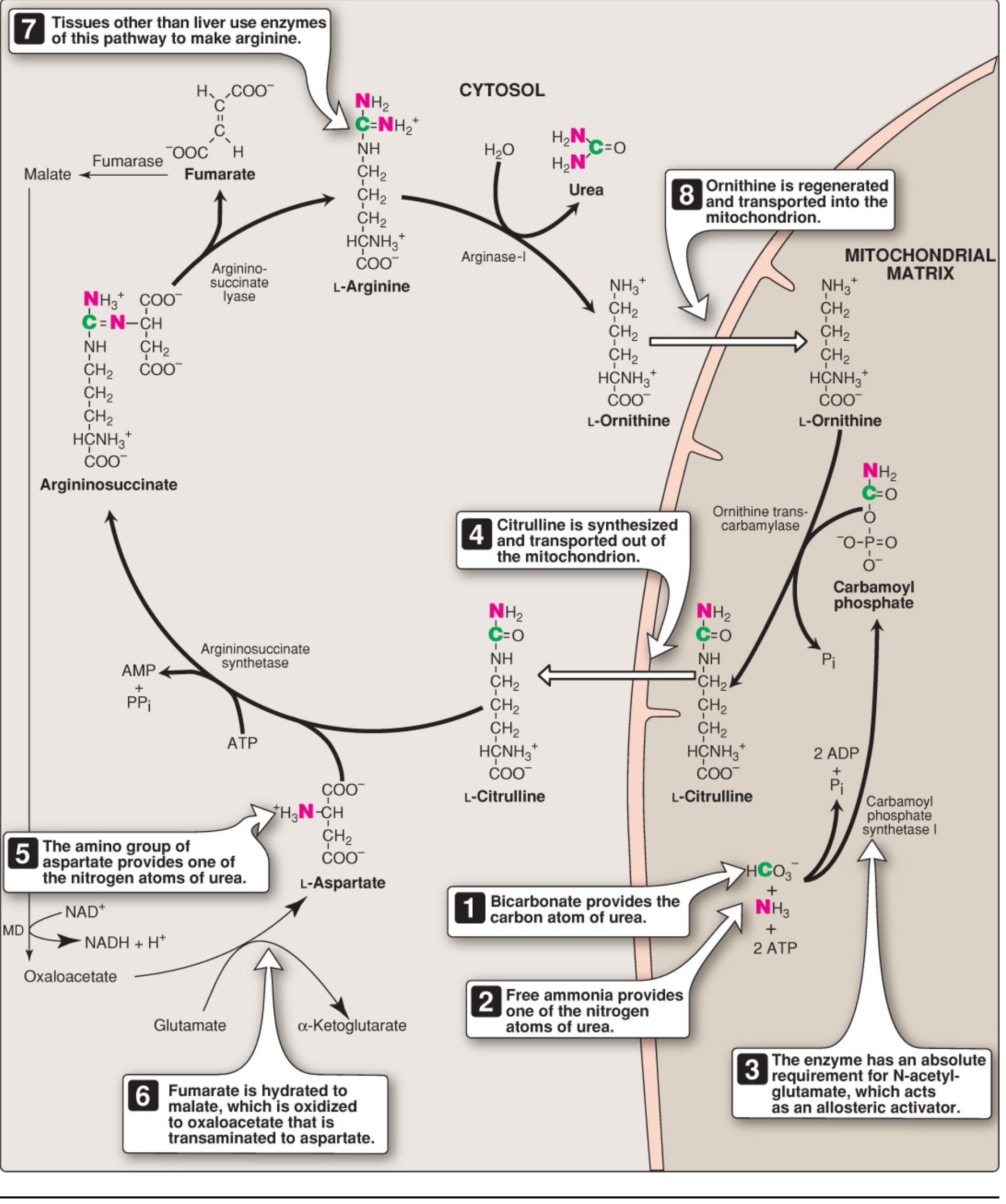
The Urea Cycle (Ornithine Cycle)
Overview
The urea cycle is the primary pathway for disposing of nitrogen from amino acid catabolism. Urea (H₂N-CO-NH₂) accounts for ~90% of the nitrogen-containing components of urine. It was first described in 1932 by Hans Krebs and Kurt Henseleit - hence the original name, the Krebs-Henseleit cycle.
- One nitrogen comes from free NH₃ (ammonia)
- The other nitrogen comes from aspartate
- The carbon and oxygen come from CO₂ (as HCO₃⁻)
- The cycle occurs in hepatocytes of the liver
- Steps 1-2 occur in the mitochondrial matrix; steps 3-5 occur in the cytosol
Urea is released into the blood (measured as BUN - blood urea nitrogen) and excreted by the kidneys.
The 5 Reactions
Step 1 - Carbamoyl Phosphate Synthesis (Mitochondria)
Enzyme: Carbamoyl phosphate synthetase I (CPS I)
NH₃ + HCO₃⁻ + 2 ATP → Carbamoyl phosphate + 2 ADP + Pi
- Requires cleavage of 2 ATP
- Requires N-acetylglutamate (NAG) as an obligatory allosteric activator
- This is the rate-limiting, committed step
- Note: CPS II (cytosolic) is a different enzyme that participates in pyrimidine synthesis; it uses glutamine as nitrogen source and does not require NAG
Step 2 - Citrulline Formation (Mitochondria)
Enzyme: Ornithine transcarbamylase (OTC)
Carbamoyl phosphate + Ornithine → Citrulline + Pi
- The carbamoyl group is transferred to ornithine
- Citrulline exits the mitochondria via an antiporter (exchanges with ornithine)
- OTC deficiency is the most common urea cycle disorder (X-linked)
Step 3 - Argininosuccinate Formation (Cytosol)
Enzyme: Argininosuccinate synthetase (ASS)
Citrulline + Aspartate + ATP → Argininosuccinate + AMP + PPi
- This step introduces the second nitrogen (from aspartate)
- Requires ATP hydrolysis to AMP + PPi (equivalent to 2 ATP)
Step 4 - Cleavage of Argininosuccinate (Cytosol)
Enzyme: Argininosuccinate lyase (ASL)
Argininosuccinate → Arginine + Fumarate
- Arginine retains both nitrogens
- Fumarate is released - it enters the TCA cycle (connects urea cycle to TCA), eventually being converted back to oxaloacetate → aspartate via transamination (the "aspartate-argininosuccinate shunt")
Step 5 - Urea Release (Cytosol)
Enzyme: Arginase I
Arginine + H₂O → Urea + Ornithine
- Urea is released and excreted
- Ornithine is regenerated and transported back into the mitochondria to restart the cycle (analogous to oxaloacetate in the TCA cycle)
Energy Cost
| Step | ATP equivalent used |
|---|---|
| CPS I (Step 1) | 2 ATP → 2 ADP + Pi |
| ASS (Step 3) | ATP → AMP + PPi (= 2 ATP equivalent) |
| Total | 4 ATP per urea molecule |
Regulation
The key regulatory enzyme is CPS I, controlled by:
- N-acetylglutamate (NAG) - obligatory allosteric activator
- NAG is synthesized from acetyl-CoA + glutamate by NAG synthase
- Arginine activates NAG synthase - so when arginine (and therefore protein load) is high, the cycle speeds up
- High protein intake and prolonged fasting both increase urea cycle enzyme expression
Link to the TCA Cycle
The fumarate released in Step 4 connects to the TCA cycle:
Fumarate → Malate (fumarase) → Oxaloacetate (malate dehydrogenase) → Aspartate (transamination with glutamate)
This regenerates aspartate for Step 3, creating a cyclic interplay between the urea cycle and TCA cycle.
Nitrogen Sources - How NH₃ and Aspartate Are Generated
- Glutamate dehydrogenase (GDH): Glutamate → α-ketoglutarate + NH₃ (feeds Step 1)
- Aspartate aminotransferase (AST): Oxaloacetate + Glutamate → Aspartate + α-ketoglutarate (feeds Step 3)
- Glutamate is therefore the immediate precursor of both nitrogen atoms in urea
Hyperammonemia
Normal blood ammonia: 5-35 μmol/L (some sources give 30-60 μM). When liver function is compromised, levels can exceed 1,000 μmol/L.
Symptoms: Tremors, slurred speech, somnolence, vomiting, cerebral edema, coma, and death.
Causes:
- Acquired - liver disease (viral hepatitis, alcohol-induced cirrhosis, portal-systemic shunting bypasses hepatic urea synthesis)
- Inherited (urea cycle disorders):
| Disorder | Deficient Enzyme | Key Features |
|---|---|---|
| CPS I deficiency | Carbamoyl phosphate synthetase I | ↑ NH₃, no orotic acid |
| OTC deficiency | Ornithine transcarbamylase | Most common; X-linked; ↑ orotic acid (carbamoyl-P diverted to pyrimidine synthesis) |
| Citrullinemia | Argininosuccinate synthetase | ↑ citrulline |
| Argininosuccinic aciduria | Argininosuccinate lyase | ↑ argininosuccinate |
| Arginase deficiency | Arginase I | ↑ arginine; spastic diplegia |
| NAGS deficiency | N-acetylglutamate synthase | ↑ NH₃; treated with N-carbamylglutamate |
Key distinguishing point: OTC deficiency produces elevated orotic acid in urine (excess carbamoyl phosphate in the cytosol is shunted into the pyrimidine synthesis pathway), while CPS I deficiency does not.
Treatment of Hyperammonemia
- Nitrogen scavengers: Sodium benzoate (conjugates glycine → hippurate, excreted) and sodium phenylbutyrate (→ phenylacetate → conjugates glutamine → phenylacetylglutamine, excreted) - both allow nitrogen excretion without urea
- Dietary protein restriction and essential amino acid supplementation
- Arginine supplementation (except in arginase deficiency) - arginine becomes essential when the cycle is blocked
- Liver transplant for severe defects
- N-carbamylglutamate (a NAG analog) for NAGS deficiency - activates residual CPS I
Sources: Biochemistry, 8th ed. Lippincott Illustrated Reviews, p. 712-722; Basic Medical Biochemistry - A Clinical Approach, 6e, p. 1321-1326
This is a shared conversation. Sign in to Orris to start your own chat.