Teach anaemia pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive content from Robbins & Kumar Pathologic Basis of Disease (the gold-standard pathology textbook). I have enough to write a thorough, well-organized teaching response covering all major types of anaemia. Let me compile this now.
Anaemia Pathology - A Complete Teaching Guide
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.); Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine 22E; Henry's Clinical Diagnosis and Management
1. Definition & Overview
Anaemia is a reduction of the total circulating red cell mass below normal limits, resulting in decreased oxygen-carrying capacity of blood and consequent tissue hypoxia.
Clinically, it is diagnosed by reductions in:
- Haematocrit (packed red cell volume/total blood volume)
- Haemoglobin concentration (below the normal range for age and sex)
These correlate well with red cell mass unless plasma volume is altered (e.g., dehydration or fluid overload).
2. Classification
There are two complementary approaches:
A. By Mechanism (Pathophysiologic)
| Mechanism | Examples |
|---|---|
| Blood Loss | Acute trauma; chronic GI/gynaecologic bleeding |
| Increased RBC Destruction (Haemolysis) | Hereditary spherocytosis, G6PD deficiency, sickle cell disease, thalassaemia, autoimmune |
| Decreased RBC Production | Iron deficiency, megaloblastic (B12/folate), aplastic anaemia, anaemia of chronic disease |
B. By Red Cell Size (Morphologic - MCV based)
This classification, pioneered by haematologist Max Wintrobe, is the most practical clinical starting point:
| Type | MCV | Causes |
|---|---|---|
| Microcytic | <80 fL | Iron deficiency, thalassaemia, anaemia of chronic disease, sideroblastic anaemia |
| Normocytic | 80-100 fL | Aplastic anaemia, renal disease, endocrinopathy, marrow invasion, pure red cell aplasia |
| Macrocytic | >100 fL | B12/folate deficiency (oval macrocytes), alcohol, hypothyroidism, liver disease, reticulocytosis (round macrocytes) |
Key diagnostic indices:
- MCV - mean cell volume (fL)
- MCH - mean cell haemoglobin (pg)
- MCHC - mean cell Hb concentration (g/dL)
- RDW - red cell distribution width (coefficient of variation of red cell volume; elevated in mixed deficiencies)
3. Haemolytic Anaemias
All haemolytic anaemias share:
- Shortened RBC lifespan (normal = 120 days)
- Elevated erythropoietin → compensatory erythroid hyperplasia → reticulocytosis
- Accumulation of Hb degradation products (bilirubin, haemosiderin)
Extravascular vs. Intravascular Haemolysis
| Feature | Extravascular | Intravascular |
|---|---|---|
| Site | Macrophages in spleen, liver, BM | Within blood vessels |
| Cause | Reduced RBC deformability | Mechanical injury, complement fixation, toxins |
| Findings | Anaemia, splenomegaly, jaundice | Anaemia, haemoglobinaemia, haemoglobinuria, haemosiderinuria |
| Haptoglobin | Mildly reduced | Markedly reduced/absent |
| Bilirubin | Unconjugated elevated | Unconjugated elevated |
| Splenomegaly | Present | Absent |
In both types, bilirubin is unconjugated, and excess excretion into bile can lead to pigment gallstones.
3a. Hereditary Spherocytosis
- Genetics: Autosomal dominant (usually); mutations in RBC membrane skeleton proteins (ankyrin, band 4.2, alpha/beta spectrin)
- Pathogenesis: Loss of membrane surface area without corresponding volume loss → spherocyte formation → trapped and destroyed in splenic cords
- Blood smear: Spherocytes (small, dense, no central pallor), reticulocytosis
- Lab: Positive osmotic fragility test; negative DAT (Coombs)
- Features: Anaemia, splenomegaly, jaundice
- Treatment: Splenectomy (removes site of destruction)
3b. G6PD Deficiency
- Genetics: X-linked; mutations destabilize G6PD enzyme
- Pathogenesis: G6PD is essential for the hexose monophosphate shunt, which generates NADPH. NADPH maintains glutathione in reduced form, protecting RBCs from oxidant damage. Without G6PD, oxidant stress (from infections, certain drugs - primaquine, dapsone, fava beans) → oxidation of Hb → Heinz bodies (precipitated Hb) → membrane damage → haemolysis
- Blood smear: Bite cells (where macrophages have "bitten out" Heinz bodies), blister cells
- Key point: Episodic, triggered haemolysis - not chronic
3c. Sickle Cell Disease
- Genetics: Autosomal recessive; point mutation in beta-globin gene (GAG→GTG; Glu→Val at position 6) producing HbS
Pathophysiology:
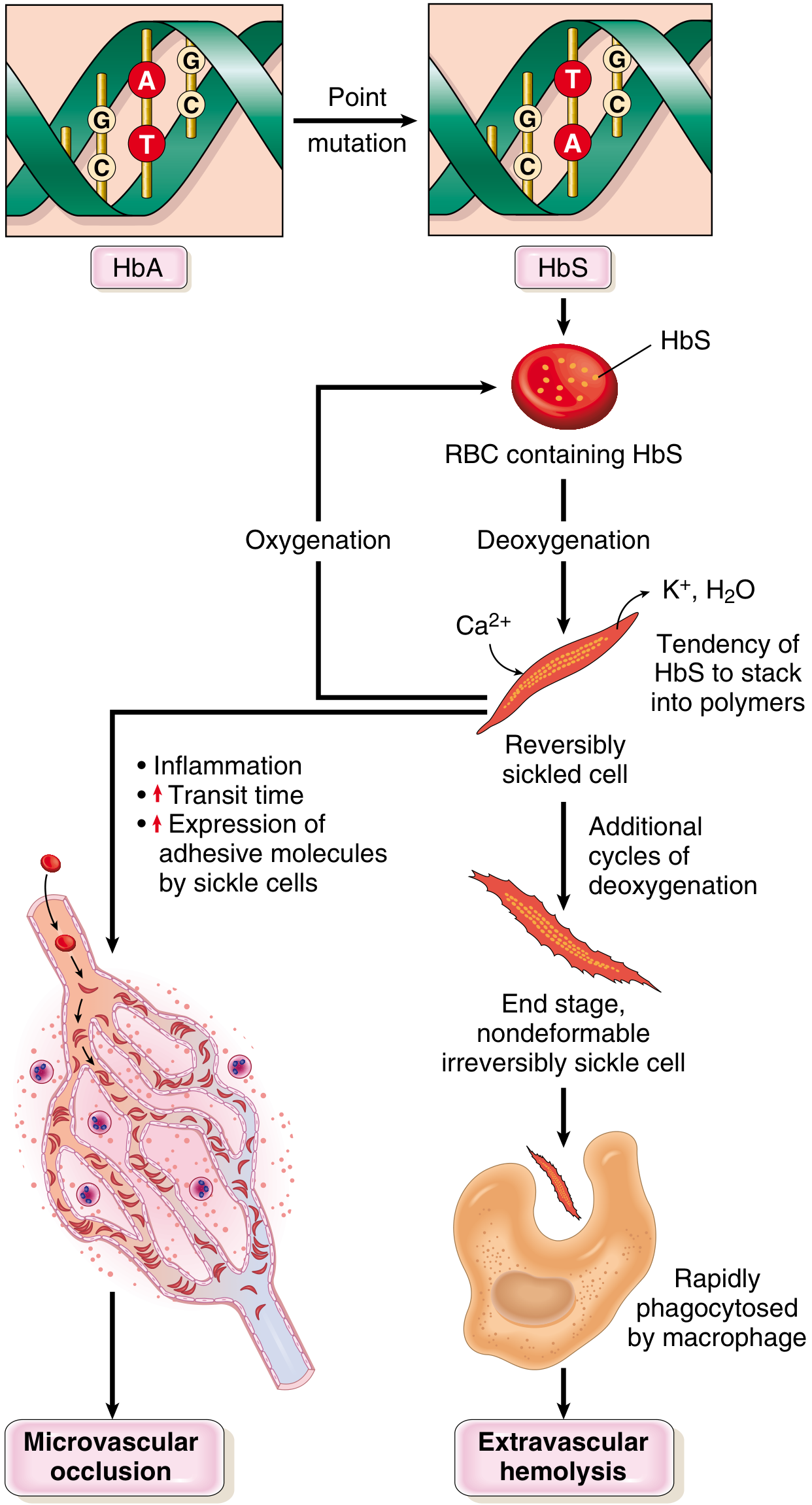
- On deoxygenation, HbS polymerizes → stacks into long rigid tactoids → distorts RBC into sickle shape
- Early sickling is reversible on reoxygenation; repeated cycles cause irreversibly sickled cells
- Sickled cells: (a) less deformable → extravascular haemolysis, (b) express increased adhesion molecules → stick to endothelium → microvascular occlusion
- Free Hb from lysed cells binds and inactivates nitric oxide → increased vascular tone + platelet aggregation → worsens vascular occlusion
Morphology:
- Peripheral smear: Irreversibly sickled cells, target cells, Howell-Jolly bodies (due to asplenia), reticulocytosis
- Bone marrow: Erythroid hyperplasia → bone resorption → "crew-cut" appearance on skull X-ray
- Spleen: Initially enlarged (red pulp congestion); progressive autosplenectomy by adolescence (fibrosis after repeated infarcts)
- Other infarcts: Bones, brain, kidney, liver, retina, pulmonary vessels (cor pulmonale)
Clinical:
- Moderate-severe haemolytic anaemia (Hct 18-30%)
- Vaso-occlusive crises - severe bone pain
- Dactylitis in children (hand-foot syndrome)
- Increased susceptibility to encapsulated organisms (due to functional asplenia) - Streptococcus pneumoniae
- HbF protects - disease is milder in early infancy
3d. Thalassaemias
- Alpha-thalassaemia: Deletion of alpha-globin genes (chromosome 16). Severity depends on number of deleted genes: 1 deletion (silent) → 4 deletions (Hb Bart's - incompatible with life)
- Beta-thalassaemia: Point mutations reducing (beta+) or eliminating (beta0) beta-globin synthesis. Excess alpha chains precipitate in RBCs → premature RBC destruction (ineffective haemopoiesis) and haemolysis
- Morphology: Microcytic hypochromic RBCs, target cells, tear-drop cells; markedly expanded marrow (maxilla prominences, "chipmunk facies", osteoporosis); extramedullary haemopoiesis (hepatosplenomegaly)
- HbA2 elevated in beta-thal trait (diagnostic)
- Treatment: Transfusion + iron chelation; stem cell transplant for severe disease
3e. Autoimmune Haemolytic Anaemia (AIHA)
Warm AIHA (37°C):
- IgG autoantibodies bind RBC surface antigens
- Opsonized RBCs phagocytosed in spleen → extravascular haemolysis
- Positive direct antiglobulin test (DAT/Coombs)
- Causes: Idiopathic, SLE, CLL, drugs (methyldopa, penicillin)
Cold Agglutinin Type (<30°C):
- IgM antibodies bind RBCs in cold peripheral vessels → agglutination + C3b deposition → extravascular haemolysis in warmer core
- Causes: Mycoplasma pneumonia (anti-I), infectious mononucleosis (anti-i), lymphoma
Cold Hemolysin Type (Paroxysmal Cold Haemoglobinuria):
- IgG Donath-Landsteiner antibodies; bind RBC P antigen in cold → complement-mediated intravascular lysis when blood warms
- Often post-viral in children
3f. Microangiopathic Haemolytic Anaemia (MAHA)
- Mechanical destruction of RBCs by narrowed microvascular lumens (thrombi in DIC, TTP, HUS, malignant hypertension)
- Blood smear: Schistocytes (helmet cells, triangle cells, burr cells)
4. Iron Deficiency Anaemia
The most common nutritional deficiency worldwide - affects ~10% in high-income and 25-50% in low-income countries.
Iron Metabolism
- Total body iron: ~2.5 g (women), ~3.5 g (men)
- 80% in functional pool (Hb, myoglobin, enzymes); 15-20% in storage pool (ferritin, haemosiderin - in macrophages of liver, spleen, BM)
- Transport: Bound to transferrin (normally ~33% saturated; serum iron ~120 µg/dL men, ~100 µg/dL women)
- Hepcidin (liver peptide): Negatively regulates ferroportin → controls iron absorption and release
- Inflammation → IL-6 → hepcidin upregulation → ferroportin degradation → iron sequestration (basis of anaemia of chronic disease)
Causes
- Inadequate intake - poverty, restricted diet, infants on cow's milk
- Malabsorption - coeliac disease, post-gastrectomy (duodenum bypassed)
- Increased requirements - pregnancy, infancy, adolescence
- Chronic blood loss (most common cause in developed countries) - GI tract (peptic ulcer, colorectal cancer, angiodysplasia), gynaecologic (menorrhagia)
- In adult males and postmenopausal females: GI blood loss must be excluded before attributing iron deficiency to any other cause - an occult GI cancer must be ruled out.
Stages of Iron Deficiency
- Depletion of iron stores - serum ferritin falls; no anaemia yet; BM iron absent (Prussian blue stain)
- Iron-deficient erythropoiesis - serum iron falls, TIBC rises, transferrin saturation <15%; no overt anaemia yet
- Iron deficiency anaemia - Hb/Hct fall; full picture of microcytic hypochromic anaemia
Morphology
Peripheral blood smear: Microcytic (small) and hypochromic (pale) RBCs - central pallor zone >1/3 of cell diameter; narrow rim of haemoglobin at periphery. Pencil cells (elongated poikilocytes). Anisocytosis and poikilocytosis.
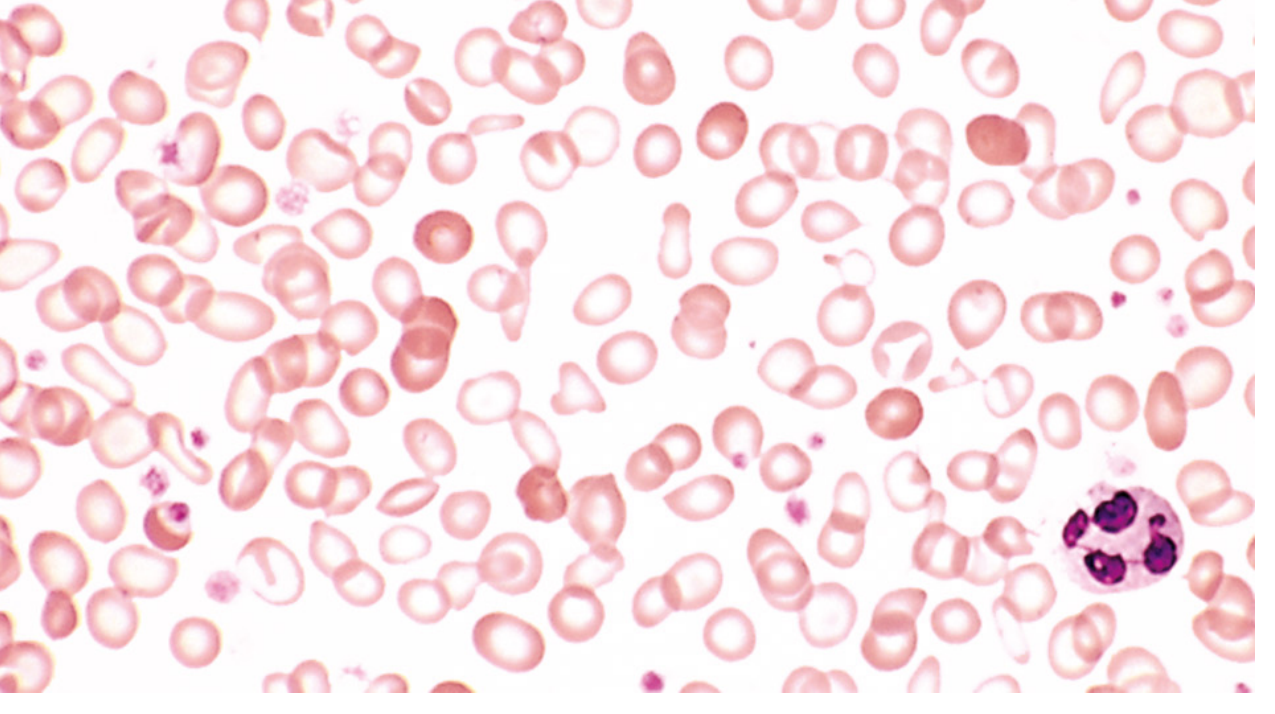
Bone marrow: Mild-moderate erythroid hyperplasia; absent stainable iron on Prussian blue stain (diagnostically significant).
Labs
| Test | Iron Deficiency | Anaemia of Chronic Disease |
|---|---|---|
| Serum iron | Low | Low |
| TIBC | High | Low/Normal |
| Transferrin saturation | Low (<15%) | Low |
| Serum ferritin | Low | High |
| BM iron stores | Absent | Increased |
Clinical Features
- Fatigue, pallor, exertional dyspnoea (general anaemia symptoms)
- Severe/chronic iron deficiency also causes:
- Koilonychia (spoon nails)
- Pica (craving for non-food items - clay, ice/pagophagia, flour)
- Angular cheilitis, glossitis (atrophic tongue)
- Alopecia, intestinal malabsorption
- In children: cognitive impairment
5. Megaloblastic Anaemia
Caused by defective DNA synthesis in erythroid (and other proliferating) cells, leading to a mismatch between nuclear maturation and cytoplasmic maturation - nuclear-cytoplasmic asynchrony.
Causes:
- Vitamin B12 (cobalamin) deficiency
- Folate deficiency
- Drugs impairing DNA synthesis (methotrexate, hydroxyurea, certain anticonvulsants)
- Myelodysplasia
Pathogenesis
- B12 and folate are both required for synthesis of thymidylate (for DNA)
- B12 deficiency traps folate in an unusable form (methyltetrahydrofolate trap)
- Result: Impaired DNA synthesis → cells grow but cannot divide → large cells with immature-appearing nuclei
Morphology
Peripheral blood:
- Macrocytic anaemia (oval macrocytes, MCV often >110 fL)
- Hypersegmented neutrophils (>5 lobes or any cell with ≥6 lobes) - classic hallmark
- Pancytopenia in severe cases
Bone marrow:
- Markedly hypercellular
- Megaloblasts - large erythroid precursors with finely dispersed ("open") nuclear chromatin despite cytoplasmic haemoglobinization
- Nuclear-cytoplasmic asynchrony (nucleus looks immature, cytoplasm is mature/haemoglobinized)
- Giant metamyelocytes and band forms
- Hypersegmented megakaryocytes
- Ineffective haemopoiesis - most precursors undergo apoptosis in marrow → pancytopenia despite hyperplastic marrow
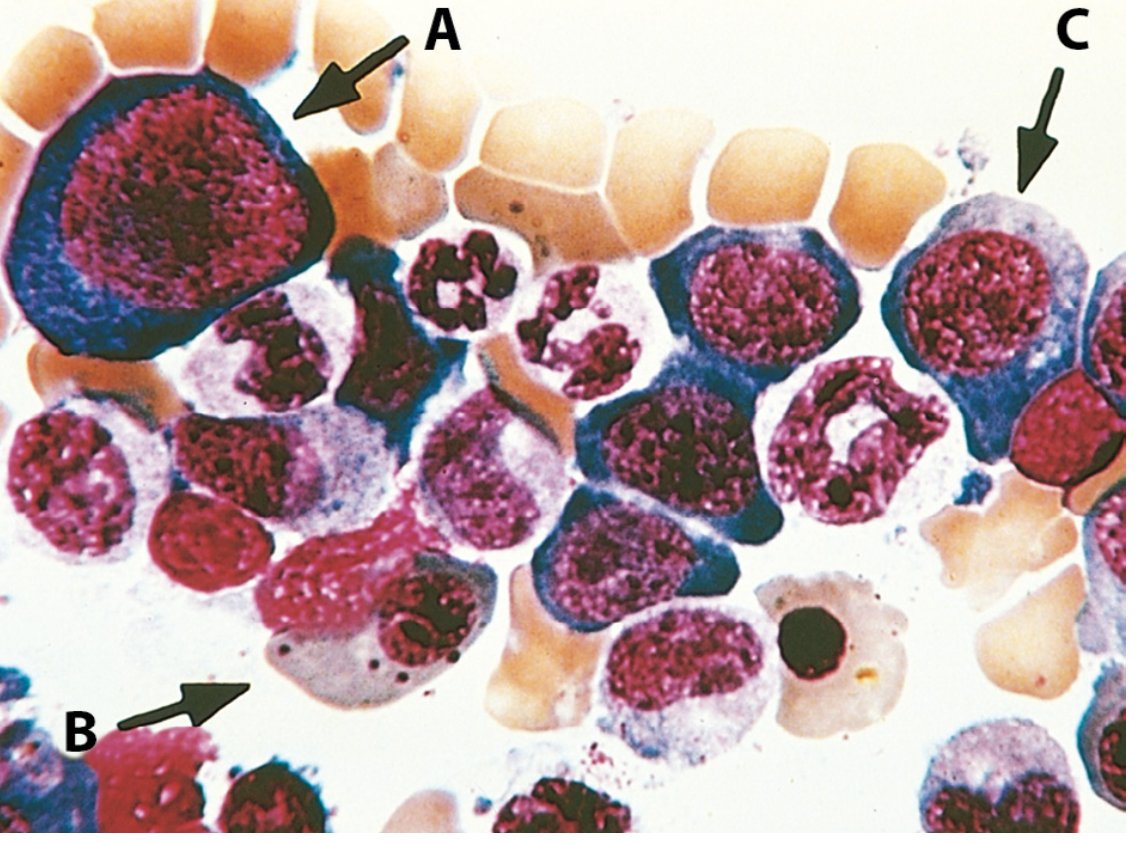
Pernicious Anaemia (B12 deficiency)
A specific form caused by autoimmune gastritis destroying parietal cells → loss of intrinsic factor (IF) → failed B12 absorption in terminal ileum.
- B12 absorption pathway: Dietary B12 freed by pepsin → binds haptocorrin in stomach → transferred to IF in duodenum → IF-B12 complex absorbed in terminal ileum via cubilin receptors
- Autoantibodies against: (1) parietal cells, (2) IF itself (blocking antibody), (3) IF-B12 complex (binding antibody)
- Associated with: Atrophic gastritis, achlorhydria, elevated gastrin
- Neurologic complication: Subacute combined degeneration of the spinal cord - demyelination of dorsal (posterior) and lateral columns → sensory ataxia, loss of vibration/proprioception, spastic paraparesis
- This neurological complication does NOT occur in folate deficiency - an important distinguishing feature
- Treatment: Intramuscular B12 injections (bypasses GI absorption)
Folate Deficiency
- Dietary sources: Green leafy vegetables, liver, nuts (destroyed by cooking)
- Daily requirement: ~100 µg; body stores last ~3-4 months only (much less than B12)
- Causes: Dietary deficiency (alcoholism, poverty), malabsorption, increased demand (pregnancy), drugs (methotrexate, trimethoprim, phenytoin)
- No neurological features (unlike B12 deficiency)
6. Aplastic Anaemia
Definition: Chronic primary haemopoietic failure with pancytopenia (anaemia + neutropenia + thrombocytopenia) due to destruction or suppression of haemopoietic stem cells.
Pathogenesis
In most cases (~80%): Autoimmune - cytotoxic T cells attack haemopoietic stem cells. This explains the response to immunosuppression.
Causes:
- Idiopathic (50-70%)
- Chemical agents:
- Dose-related (predictable): Alkylating agents, antimetabolites, benzene, chloramphenicol, inorganic arsenicals
- Idiosyncratic (unpredictable): Chloramphenicol, phenylbutazone, gold salts, carbamazepine
- Viral infections: Hepatitis (~5%), EBV, CMV, HIV
- Whole-body radiation (dose-dependent)
- Inherited: Fanconi anaemia (autosomal recessive; DNA repair defects)
Morphology
- Bone marrow: Markedly hypocellular - fatty replacement of haemopoietic cells; scattered lymphocytes and plasma cells; virtual absence of erythroid, myeloid precursors and megakaryocytes
- Peripheral blood: Pancytopenia; normocytic normochromic anaemia; no reticulocytosis (production failure)
Clinical Features
- Anaemia: Fatigue, pallor, dyspnoea
- Neutropenia: Recurrent serious infections (opportunistic organisms)
- Thrombocytopenia: Mucosal bleeding, petechiae, ecchymoses
- Prognosis: Severe aplastic anaemia is fatal without treatment
Treatment
- Young patients (<40 yrs): Allogeneic BMT (curative)
- Older patients / no matched donor: Immunosuppression (anti-thymocyte globulin + cyclosporin + eltrombopag)
- Supportive: Transfusions, growth factors, antibiotics
7. Anaemia of Chronic Disease (ACD)
Second most common anaemia after iron deficiency. Seen in:
- Chronic infections (TB, osteomyelitis, HIV)
- Chronic inflammatory conditions (rheumatoid arthritis, SLE, IBD)
- Malignancy
Pathogenesis (hepcidin-mediated):
- Inflammation → IL-6 → liver produces hepcidin
- Hepcidin degrades ferroportin on macrophages and enterocytes
- Iron is trapped in macrophage stores and cannot be released to developing erythrocytes
- Erythroid precursors are starved of iron despite adequate (or increased) body iron stores
- Additionally, cytokines suppress erythropoietin production and erythroid proliferation
Key lab distinction from iron deficiency:
- Serum ferritin: HIGH (stored iron is abundant)
- TIBC: LOW (unlike iron deficiency where TIBC is high)
- Serum iron: Low
- BM macrophage iron: Increased (Prussian blue +ve)
Blood film: Normochromic normocytic, or mildly hypochromic microcytic
8. Quick Summary Table - Distinguishing Features
| Feature | IDA | ACD | Megaloblastic | Aplastic | Haemolytic |
|---|---|---|---|---|---|
| MCV | Low | Normal/Low | High | Normal | Normal/High |
| Reticulocytes | Low | Low | Low | Very low | High |
| Serum ferritin | Low | High | Normal | Normal | Normal |
| TIBC | High | Low | Normal | Normal | Normal |
| BM | Erythroid hyperplasia, no iron | Normal, iron+ | Megaloblasts, hypercellular | Hypocellular | Erythroid hyperplasia |
| Bilirubin | Normal | Normal | Normal/raised | Normal | Unconjugated elevated |
| LDH | Normal | Normal | Elevated (ineffective eryth.) | Normal | Elevated |
| Hallmark smear | Microcytic hypochromic, pencil cells | Normal/microcytic | Oval macrocytes, hyperseg. neutrophils | Normal morphology, pancytopenia | Spherocytes / sickle cells / schistocytes |
9. Clinical Approach to Anaemia
- History: Diet, bleeding, drugs, chronic illness, family history, ethnicity
- Exam: Pallor, jaundice, splenomegaly, koilonychia, glossitis, neurological signs
- First investigations: CBC + differential, peripheral smear, reticulocyte count
- Reticulocyte count - key decision point:
- High reticulocytes → Haemopoiesis intact; problem is loss or destruction → haemolytic anaemia or blood loss
- Low reticulocytes → Production failure → iron deficiency, megaloblastic, aplastic, ACD
- MCV-directed workup:
- Low MCV → iron studies, Hb electrophoresis
- High MCV → B12, folate levels; assess for oval macrocytes + hyperseg. neutrophils
- Normal MCV → broad differential; check RFTs, TFTs, LFTs, bone marrow
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th ed. (Chapter 14)
- Robbins & Kumar Basic Pathology (Chapter 10)
- Harrison's Principles of Internal Medicine, 22nd ed. (Chapter 66)
- Henry's Clinical Diagnosis and Management by Laboratory Methods
Anemia based on mcv
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
I have comprehensive content. Let me now write the detailed MCV-based anaemia teaching guide.
Anaemia Classification by MCV
Sources: Goldman-Cecil Medicine; Henry's Clinical Diagnosis; Symptom to Diagnosis (4th ed.); Harrison's Principles of Internal Medicine 22E
The MCV Framework
MCV (Mean Corpuscular Volume) measures the average size of a red blood cell in femtoliters (fL). It is the single most useful first step in classifying anaemia because it immediately narrows the differential diagnosis.
| MCV Range | Category | Core Mechanism |
|---|---|---|
| < 80 fL | Microcytic | Defective haemoglobin synthesis |
| 80-100 fL | Normocytic | Diverse (production failure, loss, or destruction) |
| > 100 fL | Macrocytic | Defective DNA synthesis or membrane abnormality |
The reticulocyte count works alongside MCV. A high reticulocyte count (>2% corrected, or RPI >2) points to blood loss or haemolysis, regardless of MCV. A low reticulocyte count points to underproduction.
1. MICROCYTIC ANAEMIA (MCV < 80 fL)
Core Concept
All microcytic anaemias reflect impaired haemoglobin synthesis. Less Hb per cell = smaller, paler cells. The mnemonic TICS or SITA captures the main causes:
| Cause | Mechanism |
|---|---|
| Iron Deficiency | Insufficient iron for haem synthesis |
| Thalassaemia | Deficient globin chain synthesis |
| Anaemia of Chronic Disease (ACD) | Iron sequestration by hepcidin (usually normocytic, can be microcytic in long-standing disease) |
| Sideroblastic Anaemia | Defective haem synthesis despite iron availability |
| Lead Poisoning | Inhibits haem synthesis enzymes (ALA dehydratase) |
1A. Iron Deficiency Anaemia (IDA)
Most common cause of microcytic anaemia worldwide.
- Peripheral smear: Microcytic + hypochromic cells, pencil cells (elongated poikilocytes), anisocytosis, poikilocytosis
- MCV: Typically 60-79 fL
- RDW: Elevated (anisocytosis - red cells vary greatly in size)
Labs:
| Test | IDA |
|---|---|
| Serum iron | Low |
| TIBC | High (body makes more transferrin trying to capture iron) |
| Transferrin saturation | Low (<15%) |
| Serum ferritin | Low (best single test; reflects depleted stores) |
| BM iron (Prussian blue) | Absent |
1B. Thalassaemia
- Peripheral smear: Microcytic + hypochromic, target cells (codocytes), basophilic stippling, tear-drop cells
- MCV: Often very low (can be <65 fL in beta-thal major), disproportionately low compared to the haemoglobin level
- RDW: Normal or mildly elevated (cells are uniformly small, unlike IDA)
Key distinguishing feature - Mentzer Index:
Mentzer Index = MCV ÷ RBC count
- >13 → suggests Iron Deficiency (fewer, larger cells)
- <13 → suggests Thalassaemia (many small cells)
Labs:
- Serum iron: Normal or high
- Ferritin: Normal or high
- HbA2 elevated (>3.5%) → diagnostic of beta-thalassaemia trait
- HbF elevated in beta-thal major
- Hb electrophoresis is essential
1C. Anaemia of Chronic Disease (ACD)
- Usually normocytic; becomes microcytic only in long-standing disease
- Serum iron: Low
- TIBC: Low (unlike IDA - key distinguishing point)
- Ferritin: High (iron trapped in macrophages)
- Background: Chronic infection, malignancy, autoimmune disease, renal failure
1D. Sideroblastic Anaemia
- Defect in haem synthesis despite iron being present and even accumulating
- Iron loads into mitochondria around the nucleus → ring sideroblasts on Prussian blue stain of BM (pathognomonic)
- Peripheral smear: Dimorphic - two populations of red cells: one hypochromic microcytic, one normal/macrocytic → MCV may be normal or even high
Causes:
- Inherited (X-linked): Mutations in ALAS2 gene (first enzyme of haem synthesis); responds to pyridoxine (vitamin B6)
- Acquired:
- Alcohol (most common acquired cause; reversible)
- Isoniazid, chloramphenicol, lead (inhibit pyridoxal phosphate)
- Myelodysplastic syndrome (MDS with ring sideroblasts - clonal, serious)
- Copper deficiency, zinc toxicity
Labs:
- Serum iron: High (iron cannot be used)
- Ferritin: High
- TIBC: Low/normal
- Transferrin saturation: High
Distinguishing the Microcytic Anaemias
| Feature | IDA | Thalassaemia trait | ACD | Sideroblastic |
|---|---|---|---|---|
| Serum iron | Low | Normal/High | Low | High |
| TIBC | High | Normal | Low | Low/Normal |
| Ferritin | Low | Normal | High | High |
| RDW | High | Normal | Normal | High (dimorphic) |
| Mentzer (MCV/RBC) | >13 | <13 | - | - |
| Hb electrophoresis | Normal | HbA2 ↑ | Normal | Normal |
| BM iron | Absent | Normal/High | Increased | Ring sideroblasts |
| Response to iron | Yes | No | No | No (except inherited form → B6) |
2. NORMOCYTIC ANAEMIA (MCV 80-100 fL)
Core Concept
This is the broadest category. The reticulocyte count is the key next step to narrow the differential:
Normocytic Anaemia
│
├── Reticulocyte count HIGH (RPI >2)
│ → Haemolysis or Acute blood loss
│
└── Reticulocyte count LOW (RPI <2)
→ Underproduction
├── Anaemia of Chronic Disease
├── Renal disease (↓ EPO)
├── Aplastic anaemia
├── Endocrine (hypothyroidism, Addison's, hypogonadism)
├── BM infiltration (malignancy, granulomas)
└── Early iron deficiency
2A. Anaemia of Chronic Disease / Anaemia of Inflammation
- Most common normocytic anaemia
- Mild, insidious; dominated by symptoms of the underlying disease
- Normochromic normocytic (occasionally microcytic)
- Low reticulocyte count
2B. Anaemia of Chronic Kidney Disease (CKD)
- Deficient erythropoietin (EPO) production from the damaged kidney
- Normochromic normocytic; echinocytes (burr cells) occasionally seen
- Reticulocyte count low for degree of anaemia
- EPO levels inappropriately low relative to the degree of anaemia
- Treatment: Recombinant EPO (epoetin/darbepoetin) + iron supplementation
2C. Aplastic Anaemia
- Destruction of haemopoietic stem cells → pancytopenia
- Normocytic normochromic anaemia, very low reticulocytes
- BM: Markedly hypocellular (fatty marrow)
- WBC and platelet count also low
2D. Haemolytic Anaemias (Normocytic/Macrocytic)
High reticulocyte count + normocytic (or slightly macrocytic due to reticulocytosis):
| Feature | Extravascular | Intravascular |
|---|---|---|
| Splenomegaly | Present | Absent |
| Haptoglobin | Mildly reduced | Markedly reduced |
| Haemoglobinuria | Absent | Present |
| LDH | Elevated | Markedly elevated |
| DAT (Coombs) | Positive in AIHA | Positive in AIHA |
Blood smear clues in normocytic haemolytic anaemia:
- Spherocytes → Hereditary spherocytosis or warm AIHA
- Sickle cells → Sickle cell disease
- Schistocytes (helmet/triangle cells) → Microangiopathic haemolytic anaemia (DIC, TTP, HUS)
- Bite cells, blister cells → G6PD deficiency
- Target cells → Haemoglobinopathy, liver disease
2E. Endocrine Causes
- Hypothyroidism: Usually normocytic (can be macrocytic)
- Adrenal insufficiency (Addison's): Normocytic
- Hypogonadism (androgen deficiency): Androgens stimulate EPO; their deficiency causes mild normocytic anaemia
3. MACROCYTIC ANAEMIA (MCV > 100 fL)
Core Concept
Divided into two major groups based on whether hypersegmented neutrophils are present:
Macrocytic Anaemia
│
├── MEGALOBLASTIC (hypersegmented neutrophils PRESENT)
│ ├── Vitamin B12 deficiency
│ ├── Folate deficiency
│ └── Drugs (methotrexate, hydroxyurea, AZT, phenytoin)
│
└── NON-MEGALOBLASTIC (hypersegmented neutrophils ABSENT)
├── Alcohol
├── Liver disease
├── Hypothyroidism
├── Myelodysplastic syndrome (MDS)
└── Reticulocytosis (reticulocytes are larger than mature RBCs)
3A. Megaloblastic Anaemia - B12 and Folate Deficiency
Mechanism: Defective DNA synthesis → cells grow but cannot divide → nuclear-cytoplasmic asynchrony → large, immature-looking cells (megaloblasts in BM; oval macrocytes in blood)
Peripheral smear hallmarks:
- Oval macrocytes (not round)
- Hypersegmented neutrophils (≥5 lobes, or any with ≥6 lobes) - the single most specific finding
Bone marrow:
- Hypercellular
- Megaloblasts with finely open (non-pyknotic) chromatin despite haemoglobinisation
- Giant metamyelocytes
- Ineffective haemopoiesis → pancytopenia despite hyperplastic marrow
Distinguishing B12 from Folate:
| Feature | B12 Deficiency | Folate Deficiency |
|---|---|---|
| Neurological | YES - subacute combined degeneration of cord (posterior + lateral column demyelination: sensory ataxia, loss of vibration/proprioception, spastic paraparesis) | NO |
| Stores last | 3-5 years | 3-4 months |
| Serum level | B12 low (<200 pg/mL) | Folate low |
| RBC folate | Low (B12 deficiency traps folate) | Low |
| MMA (methylmalonic acid) | High | Normal |
| Homocysteine | High | High |
| Causes | Pernicious anaemia, gastrectomy, terminal ileal disease (Crohn's), veganism, fish tapeworm | Dietary (alcoholism, poverty), malabsorption, drugs, pregnancy, haemolysis |
Giving folate to a B12-deficient patient corrects the blood picture but NOT the neurological damage - always check B12 before treating with folate alone.
3B. Non-Megaloblastic Macrocytic Anaemia
No hypersegmented neutrophils. Round macrocytes (not oval).
| Cause | Mechanism | Clues |
|---|---|---|
| Alcohol | Direct toxic effect on erythropoiesis; also causes folate deficiency | History of alcohol use, raised GGT, macrocytosis may precede anaemia |
| Liver disease | Abnormal lipid loading of RBC membrane → larger surface area | Jaundice, target cells, raised transaminases |
| Hypothyroidism | Slowed erythropoiesis | Fatigue, cold intolerance, TSH elevated |
| MDS | Clonal stem cell disorder; dysplastic haemopoiesis | Pancytopenia, dysplastic cells on smear, often in elderly |
| Reticulocytosis | Reticulocytes are larger (have residual RNA) → raise MCV | High reticulocyte count from haemolysis or bleeding |
| Drugs | Hydroxyurea, AZT, azathioprine, chemotherapy | Drug history |
4. Practical Diagnostic Algorithm
ANAEMIA CONFIRMED (low Hb/Hct)
│
▼
┌─────────────────────┐
│ PERIPHERAL SMEAR │
│ + MCV + Retics │
└─────────────────────┘
│
┌─────┴──────────────────────┐
│ │
MCV <80 MCV ≥80
(MICROCYTIC) (Normal/Macro)
│ │
▼ Reticulocytes?
Iron studies │
Ferritin, TIBC ┌─────┴─────┐
Hb electrophoresis HIGH LOW
BM if needed │ │
Haemolysis Production failure
/ Blood loss │
MCV 80-100:
ACD, renal, aplastic, endocrine
MCV >100:
B12/folate vs non-megaloblastic
(look for hyperseg. neutrophils)
5. Summary - MCV-Based Differential at a Glance
| Anaemia Type | MCV | Retics | RDW | Key Smear Finding | Confirmatory Test |
|---|---|---|---|---|---|
| Iron Deficiency | Low | Low | High | Microcytic hypochromic, pencil cells | Ferritin low, TIBC high |
| Thalassaemia trait | Low | Normal | Normal | Target cells, basophilic stippling | Hb electrophoresis (HbA2 ↑) |
| Sideroblastic | Low/Normal | Low | High | Dimorphic population | BM ring sideroblasts |
| ACD | Normal (low) | Low | Normal | Normal morphology | Ferritin high, TIBC low |
| Aplastic | Normal | Very low | Normal | Pancytopenia, normal RBC morphology | BM biopsy (hypocellular) |
| Renal | Normal | Low | Normal | Burr cells (echinocytes) | Creatinine, EPO level low |
| Haemolytic | Normal/High | High | Variable | Spherocytes / sickle / schistocytes | DAT, haptoglobin, LDH |
| B12/Folate (Megaloblastic) | High | Low | High | Oval macrocytes, hyperseg. neutrophils | Serum B12, folate, MMA |
| Alcohol/Liver | High | Normal | Normal | Round macrocytes, target cells | LFTs, GGT, alcohol history |
| MDS | High | Low | High | Dysplastic cells, pancytopenia | BM biopsy + cytogenetics |
Sources:
- Goldman-Cecil Medicine, International Edition (Table 144-9)
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Symptom to Diagnosis: An Evidence-Based Guide, 4th ed.
- Harrison's Principles of Internal Medicine, 22nd ed.
Etiological classification
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
I have the complete Robbins table and Harrison's framework. I now have everything needed for a comprehensive etiological classification.
Etiological Classification of Anaemia
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.); Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine 22E
The Three Fundamental Mechanisms
Every anaemia can be traced to one (or more) of three root causes:
ANAEMIA
│
┌───────────────┼───────────────┐
│ │ │
BLOOD LOSS HAEMOLYSIS DECREASED
(↑ Destruction) PRODUCTION
(↓ Output)
In some diseases both decreased production and increased destruction contribute simultaneously - for example, thalassaemia (ineffective erythropoiesis + haemolysis) and sickle cell disease (haemolysis + vaso-occlusion).
CATEGORY 1: BLOOD LOSS
Blood loss depletes the circulating red cell mass without any intrinsic RBC defect or marrow problem. The bone marrow is intact and responds vigorously.
1A. Acute Blood Loss
| Sub-type | Examples |
|---|---|
| External haemorrhage | Trauma, surgery, lacerations |
| Internal haemorrhage | Haemothorax, haemoperitoneum, retroperitoneal bleed, ruptured aortic aneurysm |
| GI bleed | Peptic ulcer haemorrhage, oesophageal varices, Mallory-Weiss tear |
Pathophysiology:
- Plasma and RBCs lost in equal proportion → Hb/Hct do NOT fall immediately
- Over hours to days, interstitial fluid shifts into intravascular space → haemodilution → Hb/Hct fall, revealing true extent of loss
- Compensatory reticulocytosis peaks at 5-7 days
- Acutely: normocytic normochromic; after reticulocyte response: may show mild macrocytosis (reticulocytes are larger)
1B. Chronic Blood Loss
| Sub-type | Examples |
|---|---|
| GI tract | Colorectal carcinoma, peptic ulcer, angiodysplasia, IBD, hookworm infestation |
| Gynaecologic | Menorrhagia, uterine fibroids, endometriosis |
| Urinary | Haematuria (renal cell carcinoma, bladder carcinoma) |
| Respiratory | Haemoptysis (chronic) |
| Iatrogenic | Repeated blood sampling (ICU patients), haemodialysis |
Pathophysiology:
- Iron is continuously lost with each bleed (Hb contains ~0.5 mg iron/mL blood)
- Over time, iron stores are depleted → Iron Deficiency Anaemia develops (microcytic hypochromic)
- In adult males and postmenopausal women: chronic blood loss = GI malignancy until proven otherwise
CATEGORY 2: INCREASED RED CELL DESTRUCTION (HAEMOLYTIC ANAEMIAS)
Premature destruction of RBCs (normal lifespan = 120 days). The marrow attempts to compensate → reticulocytosis. Classified by location of defect (inside or outside the RBC) and by whether inherited or acquired.
Broad Sub-classification
HAEMOLYTIC ANAEMIA
│
├── INTRINSIC (Intracorpuscular) - defect within the RBC itself
│ ├── Inherited
│ └── Acquired
│
└── EXTRINSIC (Extracorpuscular) - normal RBC destroyed by outside forces
└── Always acquired
2A. Intrinsic / Intracorpuscular Defects
i. Membrane Disorders (Inherited)
| Disorder | Defect | Mechanism |
|---|---|---|
| Hereditary Spherocytosis (HS) | Ankyrin, spectrin, band 4.2 mutations | Membrane skeleton weakened → membrane blebs lost → spherocyte → trapped in splenic cords |
| Hereditary Elliptocytosis | Spectrin mutations | Elliptical RBCs; usually mild haemolysis |
| Abetalipoproteinaemia | Abnormal membrane lipids | Acanthocytes (spur cells) |
| Severe hepatocellular liver disease | Abnormal lipid loading of membrane | Spur cells (acanthocytes) |
ii. Enzyme Deficiencies (Inherited)
| Pathway | Enzyme | Deficiency |
|---|---|---|
| Hexose Monophosphate Shunt | G6PD | G6PD deficiency (X-linked) - oxidant-induced episodic haemolysis; Heinz bodies, bite cells |
| Hexose Monophosphate Shunt | Glutathione synthetase | Rare |
| Glycolytic Pathway | Pyruvate kinase (PK) | PK deficiency - chronic non-spherocytic haemolytic anaemia; echinocytes |
| Glycolytic Pathway | Hexokinase | Very rare |
G6PD deficiency is the most common enzyme defect. Triggers: primaquine, dapsone, nitrofurantoin, infections, fava beans. X-linked → primarily affects males.
iii. Haemoglobin Abnormalities (Inherited)
| Type | Disorder | Defect |
|---|---|---|
| Structurally abnormal globin (Haemoglobinopathies) | Sickle cell disease (HbS) | Glu→Val substitution at β-globin position 6; HbS polymerizes on deoxygenation → sickling, haemolysis + vaso-occlusion |
| HbC disease | Glu→Lys at β-globin position 6; target cells, mild haemolysis | |
| Unstable haemoglobins | Various mutations; Heinz body haemolytic anaemia | |
| Deficient globin synthesis (Thalassaemias) | Alpha-thalassaemia | Deletion of α-globin genes; excess β chains precipitate |
| Beta-thalassaemia | Point mutations in β-globin gene; excess α chains precipitate → ineffective erythropoiesis + haemolysis |
iv. Acquired Membrane Defect
| Disorder | Defect | Feature |
|---|---|---|
| Paroxysmal Nocturnal Haemoglobinuria (PNH) | Acquired somatic mutation in PIG-A gene → loss of GPI-anchored complement regulatory proteins (CD55, CD59) on RBC surface | Complement-mediated intravascular haemolysis; venous thrombosis; pancytopenia; haemoglobinuria (classically nocturnal) |
2B. Extrinsic / Extracorpuscular Causes
i. Antibody-Mediated (Immune Haemolysis)
| Type | Antibody | Trigger | Mechanism |
|---|---|---|---|
| Alloimmune - Transfusion reaction | IgM (ABO) / IgG | Mismatched blood transfusion | Complement-mediated intravascular lysis (IgM-ABO) or extravascular (IgG) |
| Alloimmune - Haemolytic disease of newborn (HDN) | Maternal IgG anti-D | Rh-incompatible pregnancy | IgG crosses placenta → opsonises fetal RBCs → extravascular haemolysis → hydrops fetalis |
| Warm AIHA | IgG (reacts at 37°C) | Idiopathic, SLE, CLL, drugs (methyldopa) | IgG coats RBCs → phagocytosed in spleen → extravascular haemolysis |
| Cold Agglutinin Disease | IgM (reacts <30°C) | Mycoplasma pneumonia, EBV, lymphoma | IgM agglutinates RBCs + deposits C3b in cold extremities → extravascular haemolysis when warmed |
| Paroxysmal Cold Haemoglobinuria | IgG Donath-Landsteiner (anti-P) | Post-viral in children | Biphasic: binds in cold, lysis in warm → intravascular haemolysis |
| Drug-induced | Various | Penicillin (hapten), methyldopa (autoimmune), cephalosporins (non-specific adsorption) | Positive DAT; extravascular or intravascular depending on mechanism |
ii. Mechanical Trauma
| Type | Mechanism | Condition |
|---|---|---|
| Microangiopathic Haemolytic Anaemia (MAHA) | Fibrin strands / platelet thrombi in microcirculation shear passing RBCs | DIC, TTP, HUS, malignant hypertension, pre-eclampsia (HELLP), disseminated malignancy |
| Macroangiopathic | Turbulence and pressure across defective cardiac valves | Prosthetic mechanical heart valves, severe aortic stenosis |
| Repetitive physical trauma | Direct external trauma | March haemoglobinuria (soldiers), marathon running, bongo drumming, karate |
Schistocytes (helmet cells, triangle cells, burr cells) on peripheral smear = hallmark of mechanical haemolysis.
iii. Infections of Red Cells
| Organism | Mechanism |
|---|---|
| Plasmodium falciparum (Malaria) | Intracellular parasite destroys RBCs; also immune-mediated bystander destruction |
| Babesia | Intraerythrocytic parasite; "Maltese cross" (tetrad) on smear |
| Clostridium perfringens | Bacterial exotoxins (lecithinases) digest RBC membranes → massive intravascular haemolysis |
| Bartonella | Attaches to RBC surface |
iv. Toxic / Chemical Injury
| Agent | Mechanism |
|---|---|
| Snake venoms, spider venoms | Direct membrane lysis |
| Arsenic, copper (Wilson's disease) | Oxidative membrane damage |
| Dapsone, phenazopyridine | Oxidant haemolysis (Heinz body formation), especially in G6PD deficiency |
| Hypotonic IV fluids | Osmotic lysis |
v. Sequestration
| Condition | Mechanism |
|---|---|
| Hypersplenism | Overactive spleen traps and destroys normal RBCs (and platelets/WBCs) → splenomegaly + pancytopenia; seen in portal hypertension, haematological malignancies, storage disorders |
CATEGORY 3: DECREASED RED CELL PRODUCTION (AREGENERATIVE ANAEMIAS)
The bone marrow fails to produce enough RBCs. Characterised by low reticulocyte count (reticulocytopenia) relative to the degree of anaemia.
3A. Inherited / Genetic Defects
Stem Cell Depletion
| Disorder | Defect |
|---|---|
| Fanconi Anaemia | Autosomal recessive; DNA repair gene mutations (FANC genes); progressive aplastic anaemia + congenital anomalies + predisposition to AML/squamous cell carcinoma |
| Telomerase mutations (Dyskeratosis congenita) | Short telomeres → premature stem cell exhaustion → aplasia |
| Diamond-Blackfan Anaemia | Inherited pure red cell aplasia; ribosomal protein gene mutations; present in infancy |
Defects Affecting Erythroblast Maturation
| Disorder | Feature |
|---|---|
| Thalassaemia syndromes | Ineffective erythropoiesis (cells undergo apoptosis in marrow before maturation) + peripheral haemolysis |
| Congenital dyserythropoietic anaemias (CDA) | Rare; bizarre multinucleated erythroblasts |
3B. Nutritional Deficiencies
| Nutrient | Anaemia Type | Mechanism |
|---|---|---|
| Iron | Microcytic hypochromic | Iron required for haem synthesis; deficiency → impaired Hb production |
| Vitamin B12 (Cobalamin) | Macrocytic megaloblastic | Required for thymidylate synthesis (DNA); deficiency → nuclear maturation arrest, nuclear-cytoplasmic asynchrony |
| Folate | Macrocytic megaloblastic | Same pathway as B12; stores deplete faster (~3-4 months) |
| Vitamin B6 (Pyridoxine) | Sideroblastic | Required for the first step of haem synthesis (ALA synthase) |
| Copper | Mixed (microcytic or normocytic) | Required for iron absorption and utilisation |
| Protein malnutrition | Normocytic | Reduced EPO and globin synthesis |
3C. Erythropoietin (EPO) Deficiency
| Condition | Mechanism |
|---|---|
| Chronic Kidney Disease (CKD) | Destruction of peritubular interstitial cells that produce EPO; most common cause of normocytic anaemia in CKD |
| Hypothyroidism | Reduced metabolic demand → reduced EPO production; normocytic or macrocytic |
| Hypopituitarism / Addison's | Loss of androgens and other hormones that stimulate EPO |
| Protein-calorie malnutrition | Reduced EPO |
3D. Immune-Mediated Injury of Progenitors
| Condition | Mechanism |
|---|---|
| Aplastic Anaemia | Cytotoxic T cells attack and destroy haemopoietic stem cells → pancytopenia (all three cell lines); hypocellular fatty marrow |
| Pure Red Cell Aplasia (PRCA) | Selective destruction of erythroid progenitors (BFU-E, CFU-E); WBC and platelets spared. Causes: parvovirus B19 (transient in haemolytic anaemia patients - "aplastic crisis"), thymoma, CLL, mycophenolate mofetil, recombinant EPO antibodies |
3E. Inflammation-Mediated Iron Sequestration (Anaemia of Chronic Disease / ACD)
| Step | Mechanism |
|---|---|
| 1 | Chronic infection / inflammation / malignancy → inflammatory cytokines (IL-6, IL-1, TNF) |
| 2 | IL-6 → liver produces hepcidin ↑↑ |
| 3 | Hepcidin degrades ferroportin on macrophages and gut enterocytes |
| 4 | Iron trapped in macrophage stores; cannot be delivered to erythroid precursors |
| 5 | Cytokines also suppress EPO production + directly inhibit erythroid proliferation |
| Result | Normocytic (or mildly microcytic) anaemia; iron-replete stores but iron-starved erythropoiesis |
3F. Primary Haematopoietic Neoplasms
| Disorder | Mechanism |
|---|---|
| Acute Leukaemia (AML/ALL) | Malignant blast cells crowd out normal erythroid progenitors |
| Myelodysplastic Syndrome (MDS) | Clonal stem cell disorder; dysplastic, ineffective haemopoiesis |
| Multiple Myeloma | Plasma cell infiltration of marrow; also EPO suppression |
| Myelofibrosis | Fibrosis replaces haemopoietic marrow; extramedullary haemopoiesis; teardrop cells (dacrocytes) on smear |
| Chronic Lymphocytic Leukaemia (CLL) | Marrow infiltration + often concurrent AIHA |
3G. Space-Occupying Marrow Lesions (Myelophthisic Anaemia)
Normal haemopoietic tissue replaced by abnormal infiltrating cells:
| Cause |
|---|
| Metastatic carcinoma (breast, prostate, lung, stomach most common) |
| Granulomatous infections (TB, fungal) |
| Gaucher disease and other storage disorders |
| Lymphoma |
Blood smear hallmark: Leukoerythroblastic picture - nucleated RBCs + immature myeloid cells (myelocytes, metamyelocytes) released from marrow due to architectural disruption. Teardrop cells (dacrocytes) are characteristic.
Complete Etiological Classification - Summary Tree
ANAEMIA (Etiological Classification)
│
├── 1. BLOOD LOSS
│ ├── Acute: Trauma, surgery, internal haemorrhage
│ └── Chronic: GI (carcinoma, ulcer), gynaecologic, hookworm
│
├── 2. INCREASED DESTRUCTION (HAEMOLYSIS)
│ │
│ ├── INTRINSIC (Intracorpuscular)
│ │ ├── Membrane: Hereditary spherocytosis, elliptocytosis
│ │ ├── Enzyme: G6PD deficiency, PK deficiency
│ │ ├── Hb: Sickle cell disease, thalassaemia, HbC
│ │ └── Acquired: PNH
│ │
│ └── EXTRINSIC (Extracorpuscular)
│ ├── Immune: AIHA (warm/cold), transfusion reaction, HDN, drug-induced
│ ├── Mechanical: MAHA (DIC/TTP/HUS), prosthetic valves, march
│ ├── Infections: Malaria, babesia, Clostridium
│ ├── Toxic: Venoms, arsenic, oxidant drugs
│ └── Sequestration: Hypersplenism
│
└── 3. DECREASED PRODUCTION
├── Inherited: Fanconi, Diamond-Blackfan, thalassaemia, CDA
├── Nutritional: Iron, B12, folate, B6, copper
├── EPO deficiency: CKD, hypothyroidism, hypopituitarism
├── Immune-mediated: Aplastic anaemia, PRCA (parvovirus B19)
├── Inflammation (ACD): Hepcidin ↑ → iron sequestration
├── Neoplastic: AML, MDS, myeloma, myelofibrosis, CLL
└── Marrow infiltration: Metastases, TB, storage disorders
Key Distinguishing Lab Clue: Reticulocyte Count
| Finding | Interpretation |
|---|---|
| High reticulocytes (RPI >2) | Marrow is intact and responding → Blood loss or Haemolysis |
| Low reticulocytes (RPI <2) | Marrow is failing → Decreased production |
This single result immediately places the anaemia into the correct etiological branch before any other test.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th ed. (Table 14.1) | Robbins & Kumar Basic Pathology (Table 10.1) | Harrison's Principles of Internal Medicine, 22nd ed. (Ch. 105)
This is a shared conversation. Sign in to Orris to start your own chat.