Pathophysiology of portal HTN development
Reading File
Finding Sources
Finding Sources
Searching PubMed
"portal hypertension" AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a thorough, well-illustrated answer. Let me compile it.
Pathophysiology of Portal Hypertension
Portal hypertension is defined as a pathological elevation of portal venous pressure, clinically significant when the hepatic venous pressure gradient (HVPG) exceeds 10-12 mmHg (normal <5 mmHg). An HVPG >16 mmHg carries high mortality risk, and >20 mmHg predicts treatment failure in acute variceal bleeding. - Sleisenger and Fordtran's Gastrointestinal and Liver Disease
The Core Equation
Portal pressure is governed by Ohm's law for fluids:
Portal Pressure = Portal Blood Flow × Vascular Resistance
Portal hypertension develops when resistance increases, flow increases, or both. In cirrhosis - by far the most common cause in the West - both mechanisms operate simultaneously. - Yamada's Textbook of Gastroenterology
Step 1: Increased Intrahepatic Resistance
This is the initiating event in cirrhosis. It has two components:
Fixed (Mechanical) Component - ~70%
Chronic liver injury (alcohol, viral hepatitis, NAFLD) triggers progressive fibrosis lasting typically >20 years. The consequences include:
- Collagen deposition in the space of Disse by activated hepatic stellate cells (HSCs), causing capillarization of sinusoids - loss of endothelial fenestrae and formation of a subendothelial basement membrane
- Regenerative nodule formation distorting hepatic architecture and mechanically compressing sinusoidal blood flow
- Microthrombi within sinusoids from neutrophil activation
- Sinusoidal remodeling with anastomoses between the arterial and portal systems across fibrous septa, imposing arterial pressures on the low-pressure portal venous system
This component is not immediately pharmacologically reversible, but may respond to antifibrotic therapy. - Yamada's Textbook of Gastroenterology
Dynamic (Functional) Component - ~30%
Superimposed on the fixed obstruction is active intrahepatic vasoconstriction, driven by an imbalance in vasoactive mediators:
| Decreased | Increased |
|---|---|
| Nitric oxide (NO) - endothelial dysfunction reduces eNOS activity | Endothelin-1 (ET-1) - potent vasoconstrictor |
| Prostacyclin | Angiotensinogen |
| Eicosanoids |
The NET effect is contraction of vascular smooth muscle cells and myofibroblasts, adding a dynamic, reversible layer to intrahepatic resistance. This is the component targeted by vasodilators like nitroprusside and - partially - by beta-blockers. - Robbins & Cotran Pathologic Basis of Disease, Goldman-Cecil Medicine
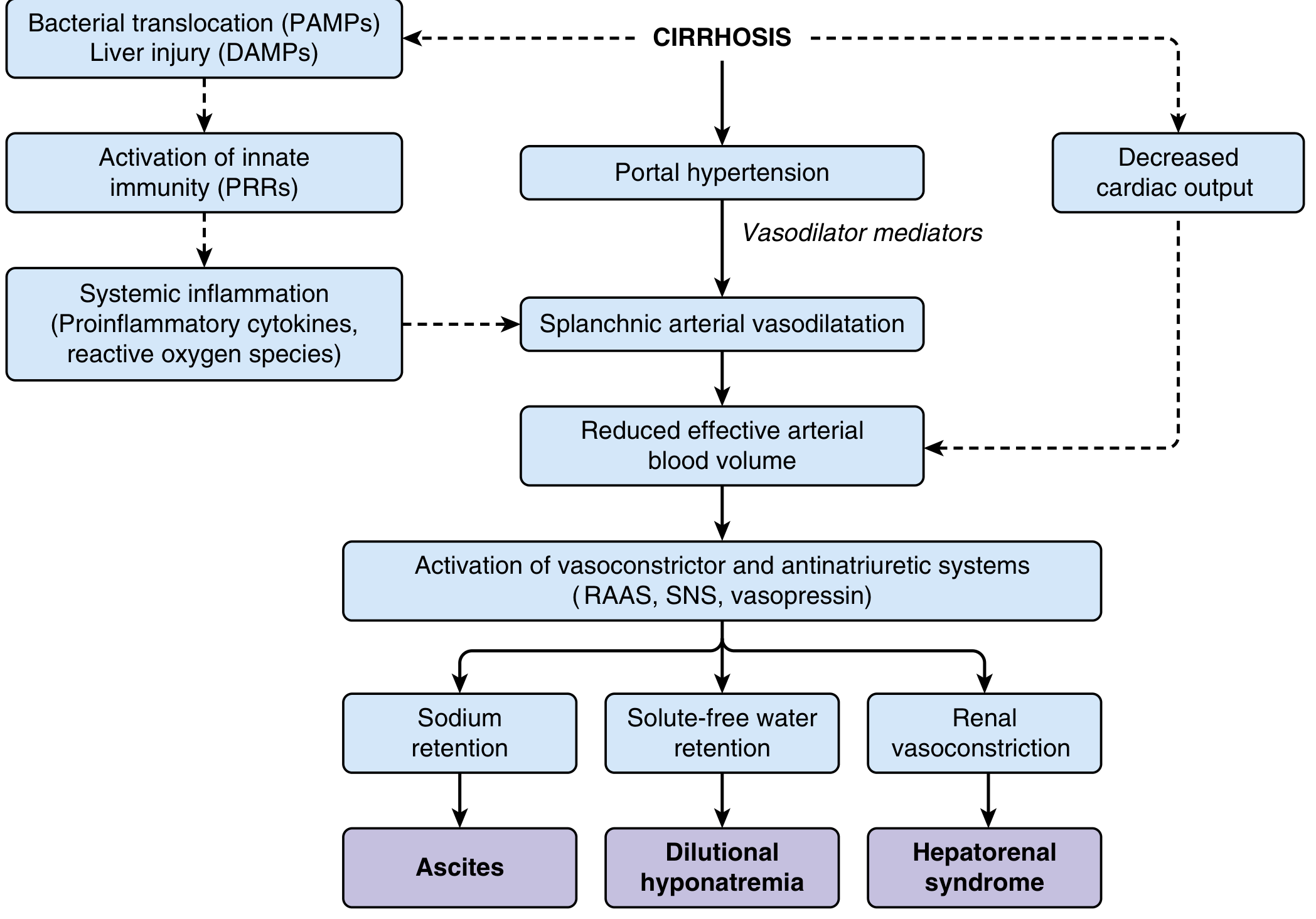
Step 2: The NO Paradox - Splanchnic Vasodilation and Increased Portal Flow
Here lies one of the most clinically important paradoxes in portal hypertension:
Intrahepatic: NO is DEFICIENT → vasoconstriction Extrahepatic/splanchnic: NO is OVERPRODUCED → vasodilation
As portal pressure rises, shear stress on splanchnic vascular endothelium upregulates eNOS (endothelial nitric oxide synthase). Bacterial translocation from the gut further amplifies NO production. Additional mediators contributing to splanchnic vasodilation include:
- Carbon monoxide (CO)
- TNF-alpha
- VEGF
- Endogenous endocannabinoids
- Prostacyclin
This splanchnic arterial vasodilation decreases vascular resistance and increases splanchnic blood flow, which drains into the portal system, elevating portal venous inflow and further increasing portal pressure. A self-perpetuating cycle is established. - Yamada's Textbook of Gastroenterology, Sabiston Textbook of Surgery
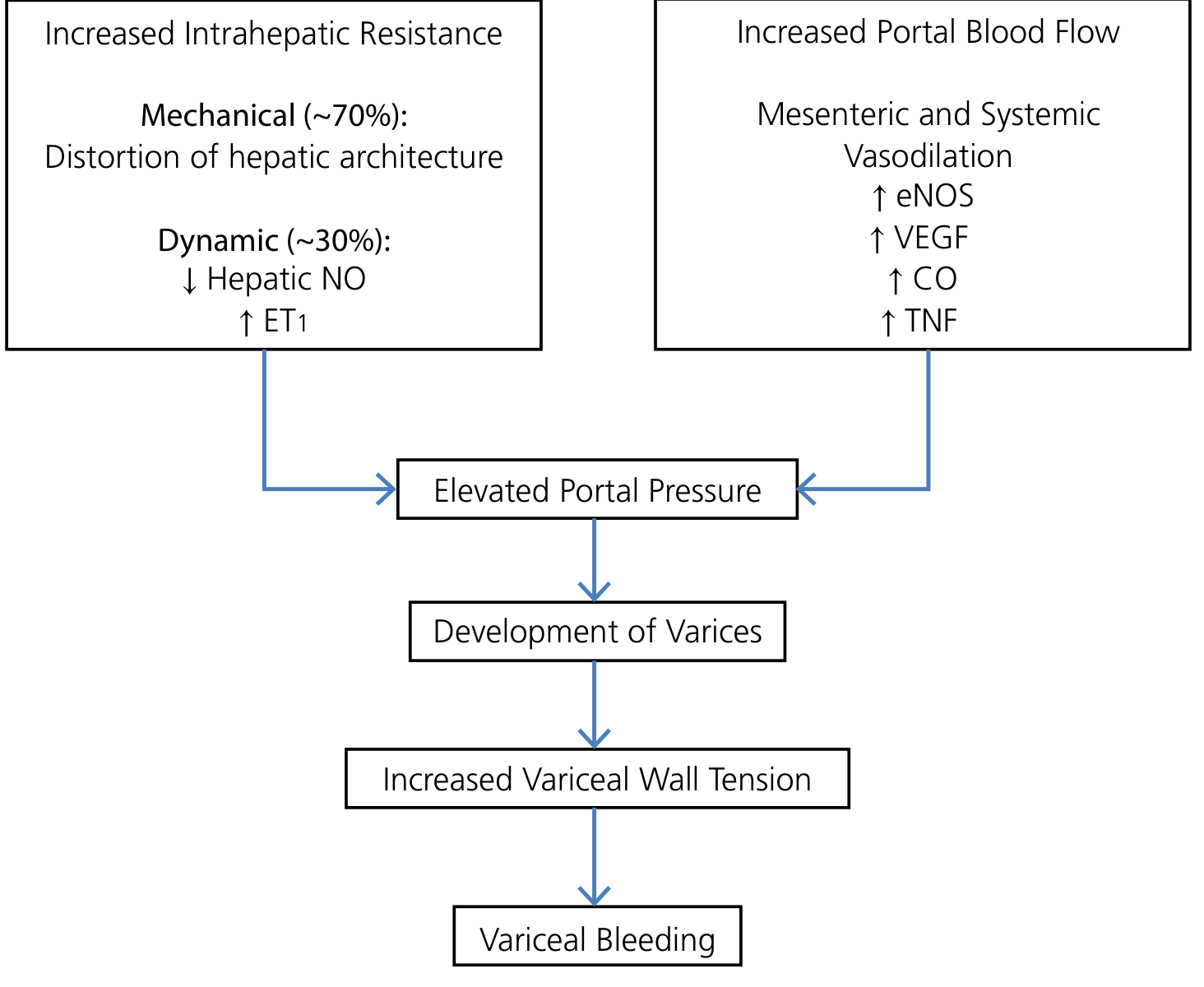
Step 3: The Hyperdynamic Circulation
Sustained splanchnic vasodilation leads to systemic arterial vasodilation and reduced effective arterial blood volume. The body responds with homeostatic neurohumoral activation:
Early Cirrhosis
- Splanchnic vasodilation is moderate
- Compensated by increased cardiac output
- Arterial pressure and volume maintained within normal limits
- Portal hypertension present but complications not yet manifest
Advanced Cirrhosis
- Splanchnic vasodilation is intense
- Reduced systemic vascular resistance cannot be compensated by further cardiac output increase
- Effective arterial hypovolemia develops
- Decreased cardiac output (cirrhotic cardiomyopathy) also contributes at this stage
- Neurohumoral activation ensues: RAAS + Sympathetic Nervous System + Vasopressin
This neurohumoral activation paradoxically maintains the hyperdynamic state by causing:
- Sodium and water retention → plasma volume expansion
- Expanded volume fills the dilated splanchnic bed → maintains the vasodilatory state
- Increased cardiac output → perpetuates high portal inflow
The net effect is a sustained hyperdynamic circulation that maintains portal hypertension even as collaterals develop. - Goldman-Cecil Medicine, Sleisenger and Fordtran's
Step 4: Portosystemic Collateral Formation
As portal pressure rises above the threshold of 10-12 mmHg HVPG, vascular channels connecting the portal venous system to the systemic venous circulation dilate and remodel (varices). Key collateral sites include:
| Site | Clinical Manifestation |
|---|---|
| Distal esophagus/proximal stomach (coronary vein) | Gastroesophageal varices - risk of catastrophic hemorrhage |
| Umbilical vein remnant | Caput medusae |
| Hemorrhoidal plexus | Rectal varices |
| Retroperitoneal and diaphragmatic veins | Miscellaneous varices |
Despite collateral formation, portal pressure is not adequately decompressed - collaterals offer additional resistance and the increased portal inflow from splanchnic vasodilation perpetuates hypertension. Variceal rupture occurs when wall tension (governed by the Law of Laplace: Tension = Pressure × Radius / Wall thickness) exceeds the tensile strength of the vessel wall. - Yamada's Textbook of Gastroenterology, Goldman-Cecil Medicine
Major Consequences - Downstream Complications
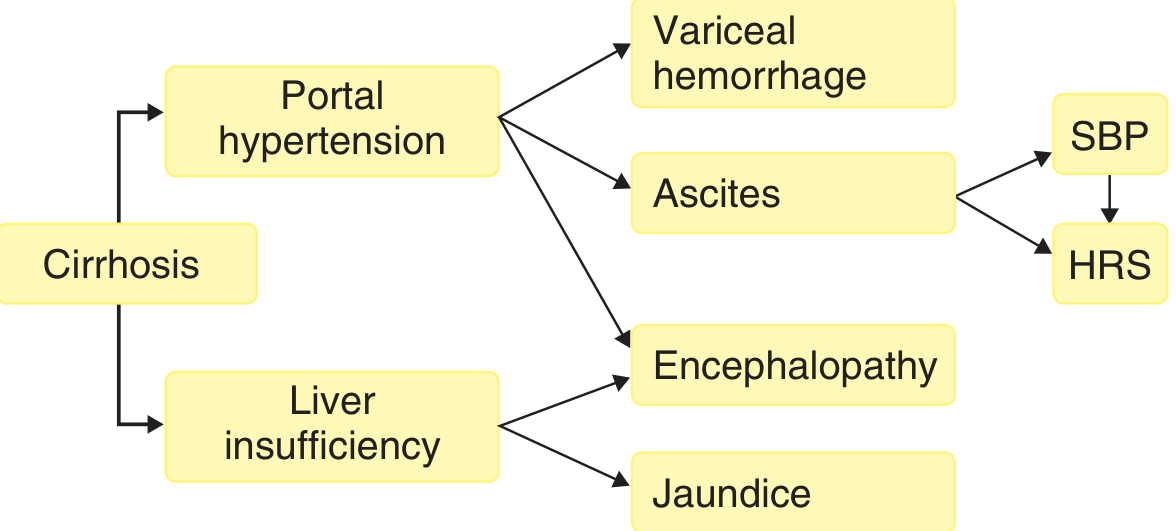
1. Ascites
Multifactorial:
- Sinusoidal hypertension increases hydrostatic pressure → fluid leaks into peritoneum
- Splanchnic vasodilation preferentially retains expanded plasma volume in the splanchnic bed
- RAAS/SNS activation causes avid Na+ and water retention
- Hypoalbuminemia reduces oncotic pressure
- Increased hepatic lymph flow overloads thoracic duct
2. Hepatic Encephalopathy
- Portosystemic shunting bypasses hepatic clearance of gut-derived toxins (ammonia, mercaptans, false neurotransmitters)
- Reduced hepatocellular mass (liver insufficiency) also contributes
3. Congestive Splenomegaly
- Elevated back-pressure in splenic vein → splenomegaly → hypersplenism
- Thrombocytopenia, leukopenia, anemia
4. Hepatorenal Syndrome (HRS)
- Extreme activation of RAAS, SNS, and vasopressin causes severe renal vasoconstriction
- Reduced GFR despite structurally intact kidneys
- Triggered by further reduction in effective arterial volume
Anatomical Classification of Causes
| Level | Examples |
|---|---|
| Prehepatic | Portal vein thrombosis, splenic vein thrombosis, portal vein narrowing, splenomegaly with increased splenic flow |
| Intrahepatic - Presinusoidal | Schistosomiasis, nodular regenerative hyperplasia, primary biliary cholangitis, sarcoidosis |
| Intrahepatic - Sinusoidal | Cirrhosis (most common in the West), massive fatty change |
| Intrahepatic - Postsinusoidal | Veno-occlusive disease (sinusoidal obstruction syndrome) |
| Posthepatic | Budd-Chiari syndrome (hepatic vein thrombosis), severe right heart failure, constrictive pericarditis |
- Robbins & Cotran Pathologic Basis of Disease
Summary of Mechanisms
Chronic Liver Injury
↓
Fibrosis + Nodule Formation
↓
↑ Fixed Intrahepatic Resistance (architectural distortion)
+
↓ Intrahepatic NO, ↑ ET-1 → ↑ Dynamic Intrahepatic Resistance
↓
Portal Hypertension (initiating event)
↓
↑ Splanchnic NO (eNOS, bacterial translocation, TNF, CO, VEGF)
↓
Splanchnic Arterial Vasodilation → ↑ Portal Inflow
↓
↑ Portal Pressure maintained/worsened (self-perpetuating cycle)
↓
Systemic vasodilation → ↓ Effective arterial blood volume
↓
RAAS + SNS + Vasopressin activation → Na/H₂O retention
↓
Hyperdynamic circulation → ↑ Cardiac output → ↑ Portal inflow
↓
Portosystemic collaterals → Varices
Ascites / Dilutional hyponatremia / Hepatorenal syndrome
Splenomegaly / Hypersplenism
Hepatic encephalopathy
Recent literature note: A 2024 Lancet Gastroenterology & Hepatology review (PMID 38642564) by Guixé-Muntet et al. provides updated mechanistic insights into cirrhotic portal hypertension, including emerging therapeutic targets such as HSC deactivation, sinusoidal endothelial cell restoration, and modulation of the gut-liver axis - consistent with the textbook framework above.
Write pathophysiology in easy language
Portal Hypertension - In Simple Language
What is the Portal Vein?
Think of the portal vein as a highway that carries blood from your intestines and spleen to your liver. Normally, blood flows through this highway at low pressure - like a quiet country road.
Portal hypertension = this highway becomes a high-pressure traffic jam.
Normal portal pressure: < 5 mmHg
Dangerous portal pressure: > 10-12 mmHg
Why Does the Pressure Build Up?
There are two main reasons, and in cirrhosis, both happen at the same time.
Reason 1: The Liver Becomes a Blocked Road
Imagine the liver is a sponge that blood normally flows through easily.
In chronic liver disease (alcohol, hepatitis, fatty liver), the liver gets scarred over years. Healthy, soft liver tissue is replaced by hard scar tissue (fibrosis). Eventually nodules of dead tissue form all over the liver (cirrhosis).
This scar tissue does two things:
A. Physical Blockage (Fixed - ~70%)
The scar physically squeezes and distorts the tiny blood channels (sinusoids) inside the liver. Blood cannot flow through freely. It is like trying to push water through a crushed pipe.
B. Active Tightening (Dynamic - ~30%)
On top of the physical blockage, the liver actively tightens its blood vessels because of a chemical imbalance:
| Normal Balance | In Cirrhosis |
|---|---|
| Nitric oxide (NO) - a natural relaxer of blood vessels | NO is LOW in the liver → vessels tighten |
| Endothelin-1 - a natural tightener | ET-1 is HIGH → vessels tighten even more |
So the liver's blood vessels clamp down, worsening the blockage. Blood piles up behind the liver → portal pressure rises.
Reason 2: Too Much Blood Rushes In (The Overflow Problem)
Here is where it gets interesting. As portal pressure rises, the body tries to compensate - but accidentally makes things worse.
The NO Paradox
Inside the liver: NO is LOW → vessels tighten Outside the liver (in the gut/splanchnic area): NO is HIGH → vessels RELAX
Why does NO go up outside the liver? Because:
- High pressure in the portal system physically stretches the gut blood vessels
- Stretching turns on an enzyme (eNOS) that makes more NO
- Gut bacteria also leak through the intestinal wall and trigger more NO production
- Other chemicals pile on: TNF, carbon monoxide, VEGF
All this extra NO causes the gut blood vessels to dilate widely (splanchnic vasodilation).
What happens when gut vessels dilate?
More blood pools in the gut. More blood drains into the portal vein. More flow into an already blocked system = even higher pressure.
It is like a traffic jam getting worse because more cars keep entering the highway from side roads.
Reason 3: The Body's "Fix" Makes Everything Worse
The wide-open gut vessels mean less blood reaches the rest of the body. The brain thinks the body is low on blood volume (even though total blood is normal - it is just pooled in the wrong place).
So the body activates its emergency systems:
Brain senses "low blood volume"
↓
Activates RAAS (Renin-Angiotensin-Aldosterone System)
Activates Sympathetic Nervous System
Releases Vasopressin
↓
Kidneys retain Salt and Water
↓
Blood volume EXPANDS
↓
Heart pumps HARDER and FASTER (hyperdynamic circulation)
↓
Even MORE blood flows into the portal system
↓
Portal pressure stays HIGH (or gets worse)
The body's attempt to fix the problem actually perpetuates the high portal pressure. This is the self-sustaining cycle of portal hypertension.
What Happens Next? - The Consequences
Because blood cannot flow forward through the liver normally, it backs up and finds alternative routes.
1. Varices (Collateral Veins)
Blood forces open small backup veins that are normally closed. These form at:
- Lower esophagus and stomach → esophageal and gastric varices
- Belly button area → caput medusae (visible veins on the belly)
- Rectum → rectal varices
These veins were never designed to carry high-pressure blood. They swell up like balloons. If they burst → life-threatening bleeding.
2. Ascites (Fluid in the Belly)
- High pressure in liver sinusoids pushes fluid out into the belly
- Retained salt and water from RAAS activation has nowhere to go except the belly
- Low albumin (from failing liver) means less fluid stays in blood vessels
- Result: fluid accumulates in the abdomen
3. Splenomegaly (Enlarged Spleen)
- Blood backs up into the spleen (it drains into the portal vein)
- Spleen enlarges and starts trapping blood cells
- Low platelets, low white cells, low red cells (hypersplenism)
4. Hepatic Encephalopathy (Brain Fog)
- Blood bypasses the liver through collateral veins
- Toxic waste (especially ammonia from gut bacteria) is not cleaned by the liver
- These toxins reach the brain → confusion, drowsiness, coma
5. Hepatorenal Syndrome (Kidney Failure)
- Extreme activation of RAAS and SNS constricts kidney blood vessels severely
- Kidneys stop filtering blood properly
- Kidney failure - even though the kidneys themselves are structurally normal
The Big Picture - One Simple Flow
Years of liver damage (alcohol / hepatitis / fat)
↓
Liver scars and stiffens → blood cannot flow through
↓
PORTAL PRESSURE RISES
↓
Gut vessels over-dilate (too much NO)
→ More blood floods the portal vein
→ Pressure rises further ←────────────┐
↓ │
Body panics: activates RAAS + SNS │
→ Retains salt & water │
→ Heart pumps more blood ───────────────┘
↓
Blood seeks escape routes
↓
┌──────────────────────────────────────┐
│ Varices → Bleeding │
│ Ascites → Distended belly │
│ Big spleen → Low blood counts │
│ Encephalopathy → Brain fog │
│ Hepatorenal syndrome → Kidney failure│
└──────────────────────────────────────┘
One Sentence Summary
The scarred liver blocks blood flow → pressure builds up behind it → the body over-dilates gut vessels to compensate → this floods the portal system with even more blood → a vicious self-reinforcing cycle develops → blood finds dangerous escape routes → multiple organ complications follow.

Expalin this
analyse_media
Your notes are about Portosystemic Collaterals - the "escape routes" blood takes when portal pressure is too high. Let me explain each one in simple language.
Formation of Varices and Complications - Portosystemic Collaterals
The Core Idea - Why Do Collaterals Form?
When portal pressure rises, blood cannot flow forward through the blocked liver. So it looks for alternative back-roads to reach the heart. These back-roads are small veins that connect the portal venous system to the systemic venous system. Normally they carry very little blood - but in portal hypertension they get flooded with high-pressure blood, swell up, and become varices.
The 4 Sites Your Notes Cover
1. Lower Esophagus
| Portal side | Left gastric vein (also called coronary vein) |
| Systemic side | Azygos vein |
| Clinical result | Esophageal varices |
Simple explanation:
Blood from the stomach/gut backs up into the left gastric vein. This connects with the azygos vein (which drains into the heart). The tiny veins at the lower end of the food pipe (esophagus) get stretched with this high-pressure blood. They balloon out into the esophageal lining → esophageal varices.
These are the most dangerous because the esophagus wall is thin. If they rupture → massive, life-threatening vomiting of blood (hematemesis). This is the #1 killer in portal hypertension.
2. Rectum
| Portal side | Superior rectal vein |
| Systemic side | Middle and Inferior rectal veins |
| Clinical result | Hemorrhoids |
Simple explanation:
The rectum (last part of the large intestine) has veins that connect the portal system (superior rectal vein) to the systemic system (middle and inferior rectal veins). High portal pressure forces blood into these connections. The rectal veins swell → hemorrhoids (piles).
Note: hemorrhoids in portal hypertension are called anorectal varices - they are different from common hemorrhoids and bleed much more severely.
3. Umbilicus (Belly Button)
| Portal side | Paraumbilical vein |
| Systemic side | Epigastric veins |
| Clinical result | Caput medusae |
Simple explanation:
There is a tiny vein near the belly button (the paraumbilical vein) that is normally closed after birth. In portal hypertension, high pressure forces this vein open again. Blood flows from the portal vein → through the paraumbilical vein → out to the epigastric veins on the belly wall.
This creates visible, tortuous (twisted) veins radiating outward from the belly button - like snakes coming out of it. This is called Caput Medusae (Latin for "head of Medusa" - the Greek monster with snakes for hair).
It is a classic sign you can see on a patient's abdomen.
4. Retroperitoneum (Behind the Belly)
| Portal side | Colic veins |
| Systemic side | Lumbar veins |
| Clinical result | Retroperitoneal shunts |
Simple explanation:
The colic veins (draining the colon) connect with lumbar veins (draining into the inferior vena cava) behind the abdominal organs. These form shunts (shortcuts) in the retroperitoneal space. They are not visible externally and rarely cause major complications, but they contribute to the general portosystemic shunting.
Visual Summary of Your Notes
PORTAL HYPERTENSION
Blood cannot go through liver
↓
Finds 4 escape routes:
SITE PORTAL VEIN → SYSTEMIC VEIN → RESULT
──────────────────────────────────────────────────────────────
Lower Esophagus Lt Gastric vein → Azygos vein → Esophageal varices ⚠️
Rectum Sup Rectal vein → Mid/Inf rectal → Hemorrhoids
Umbilicus Paraumbilical → Epigastric vein → Caput medusae
Retroperitoneum Colic vein → Lumbar vein → Retroperitoneal shunts
Why "Intra Hepatic" is Written on the Side?
The note says INTRA HEPATIC on the left margin - this refers to the cause of portal hypertension (cirrhosis is an intrahepatic cause). The collaterals shown are the consequence of that intrahepatic blockage.
Key Point to Remember
All four collateral sites share the same principle:
Portal vein (high pressure) → tiny connecting veins get stretched → drain into systemic veins (low pressure)
The bigger the pressure difference, the bigger and more dangerous the varices. Esophageal varices are the most feared because rupture = 30-50% mortality per episode.
Explain why esophageal varices bleed but not other varices
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Excellent detail from the textbooks. Here is a thorough, simple explanation:
Why Do Esophageal Varices Bleed More Than Others?
The short answer: esophageal varices are in the worst possible location, with the worst possible wall structure, exposed to the worst possible pressures.
Let me break this down using one key law of physics.
The Physics Behind It: Laplace's Law
Wall Tension = (Pressure × Radius) ÷ Wall Thickness
A vein will rupture when wall tension exceeds the strength of the vessel wall.
So three things determine whether a varix bleeds:
- Higher pressure → more tension → more likely to bleed
- Bigger radius (wider diameter) → more tension → more likely to bleed
- Thinner wall → less resistance to tension → more likely to bleed
Esophageal varices lose on all three counts compared to varices elsewhere. - Yamada's Textbook of Gastroenterology
Factor 1: Negative Intrathoracic Pressure
This is the most unique factor to esophageal varices.
The esophagus sits in the chest cavity, where the pressure is negative (below atmospheric pressure). This is what keeps your lungs inflated.
For a varix, what matters is transmural pressure = pressure inside the vein minus pressure outside it.
Transmural pressure = Intravariceal pressure - External (surrounding) pressure
In the chest (esophagus): External pressure is NEGATIVE
→ Transmural pressure = High + (minus a negative) = EVEN HIGHER
In the abdomen (rectum, umbilicus): External pressure is POSITIVE
→ Transmural pressure = High - Positive = LOWER
So the chest location actively sucks the varix outward, increasing tension on the wall. The abdomen actually helps compress veins from outside. This is the biggest single reason esophageal varices are so dangerous. - Yamada's Textbook of Gastroenterology
Factor 2: Thinnest Wall, No Support
Compare the walls at each site:
| Site | Wall Support |
|---|---|
| Esophagus | Thin mucosa only, no surrounding connective tissue, no muscle support, no perforating veins to dissipate pressure |
| Rectum | Thick rectal wall with surrounding muscle and connective tissue that physically supports the vein |
| Umbilicus (caput medusae) | Lies in the abdominal wall, surrounded by fat and connective tissue |
| Retroperitoneum | Deep, surrounded by tissue on all sides |
The esophagus has only a thin, delicate mucosal lining sitting directly over the varix. There is no connective tissue scaffold, no muscle backing, nothing to hold it together. When the vein swells, nothing stops it from bursting through. - Yamada's Textbook of Gastroenterology
Factor 3: No Perforating Veins to Release Pressure
In other parts of the body, large veins have perforating veins - small branches that act like pressure relief valves, diverting blood away before pressure gets too high.
Esophageal varices at the lower end of the esophagus lack these perforating veins. There is no escape route for the pressure. It all stays concentrated in one ballooning vessel until it bursts.
Factor 4: Mechanical Trauma From Swallowing
Every time a patient swallows food, the bolus physically passes over the varices. This is repeated mechanical trauma against an already fragile, ballooning vein. The esophagus is a mechanical organ that contracts and squeezes 24/7.
Rectal varices and caput medusae do not face this kind of repeated physical abrasion.
Factor 5: Acid Exposure (in the Lower Esophagus)
Esophageal varices form at the lower end of the esophagus, right where stomach acid can reflux upward. Acid exposure causes:
- Mucosal erosion
- Weakening of the thin overlying tissue
- Further thinning of the already fragile wall
This adds yet another mechanism of injury specific to this location.
Why Don't Hemorrhoids Bleed as Catastrophically?
Hemorrhoids do bleed - you see blood on tissue paper. But they rarely cause life-threatening hemorrhage. Why?
- They are surrounded by thick rectal wall and perianal tissue
- External pressure from surrounding tissue limits how much they can expand
- They are in the abdomen/pelvis, where positive abdominal pressure compresses the vein from outside
- Their walls have better structural support
- They are accessible and easy to compress manually
When hemorrhoids bleed massively in portal hypertension, they are technically called anorectal varices - and these CAN bleed severely, but still far less commonly than esophageal varices.
Why Don't Caput Medusae Bleed?
- They are surface veins on the abdominal wall, not internal
- Surrounded by skin, fat and connective tissue on all sides
- If they bleed, you can simply press on them from outside
- They drain in multiple directions, so pressure is dispersed
- No suction effect (no negative surrounding pressure)
Summary Table
| Factor | Esophageal | Rectal | Umbilical | Retroperitoneal |
|---|---|---|---|---|
| Surrounding pressure | Negative (chest) | Positive | Positive | Positive |
| Wall thickness | Very thin | Thick | Moderate | Moderate |
| Connective tissue support | None | Present | Present | Present |
| Perforating veins | Absent | Present | Present | Present |
| Mechanical trauma | Yes (swallowing) | Minimal | Minimal | None |
| Acid exposure | Yes | No | No | No |
| Bleeding risk | HIGHEST | Moderate | Lowest | Lowest |
One Simple Analogy
Think of each varix as a balloon:
-
Esophageal varix = A very thin balloon, inflated to maximum, being stretched from outside by a vacuum, with no support, and someone rubbing it with sandpaper (acid + food). Of course it bursts.
-
Rectal varix = A balloon inside a thick sock, with a slight pressure pushing inward from the outside. It swells, but the sock holds it.
-
Caput medusae = A balloon under your skin. It bulges out, but you can press it down with your hand.
Bottom line: Esophageal varices bleed because the physics (Laplace's law + negative intrathoracic pressure), anatomy (thin wall, no support), and environment (acid, mechanical trauma) all conspire against them simultaneously. Each episode of bleeding carries a 15-20% mortality risk - which is why prevention and prompt treatment are critical. - Robbins & Cotran Pathologic Basis of Disease, Yamada's Textbook of Gastroenterology
Causes pf portal thn
Based on what was already covered in our earlier detailed session, here is a clear, organized breakdown of all causes of portal hypertension:
Causes of Portal Hypertension
Causes are classified by where the obstruction/problem occurs - Before, Inside, or After the liver.
The Classification Framework
BLOOD FLOW: Gut → Portal Vein → LIVER → Hepatic Veins → Heart
PRE-HEPATIC INTRA-HEPATIC POST-HEPATIC
(before liver) (inside liver) (after liver)
1. PRE-HEPATIC Causes
(Block before blood even enters the liver)
The liver itself is normal. Problem is in the portal vein or spleen.
| Cause | Mechanism |
|---|---|
| Portal vein thrombosis | Blood clot blocks the portal vein - most common prehepatic cause |
| Splenic vein thrombosis | Clot in splenic vein → blood backs up (causes isolated gastric varices) |
| Portal vein compression | Tumor, lymph node, or pancreatitis pressing on the portal vein from outside |
| Portal vein narrowing/atresia | Congenital narrowing - seen in children |
| Splenomegaly with high splenic flow | Massively enlarged spleen pumps too much blood into portal vein (e.g., in malaria, kala-azar) |
| Arteriovenous fistula | Abnormal connection between splenic artery and vein → floods portal with arterial blood |
Key feature: Liver function is normal. HVPG is normal because the block is before the liver. Patients bleed from varices but tolerate it better (better liver function = better clotting).
2. INTRA-HEPATIC Causes
(Block inside the liver)
This is the most common category. Further divided by where inside the liver the block is:
A. Pre-Sinusoidal (before the sinusoids)
| Cause | Notes |
|---|---|
| Schistosomiasis | Eggs lodge in portal tracts → granulomas → fibrosis. Most common cause of portal HTN worldwide (endemic in Africa, Middle East, South America) |
| Primary biliary cholangitis (PBC) | Autoimmune destruction of bile ducts → portal tract inflammation |
| Nodular regenerative hyperplasia | Non-cirrhotic nodules compress portal venules |
| Congenital hepatic fibrosis | Genetic condition with portal fibrosis but preserved lobular architecture |
| Sarcoidosis | Granulomas in portal tracts |
| Idiopathic non-cirrhotic portal hypertension | Unknown cause, obliterative portal venopathy |
B. Sinusoidal (at the sinusoid level)
| Cause | Notes |
|---|---|
| Cirrhosis | Most common cause in the West - alcohol, HBV, HCV, NAFLD, autoimmune, Wilson's, hemochromatosis etc. |
| Massive fatty change (steatosis) | Swollen hepatocytes physically compress sinusoids |
| Acute alcoholic hepatitis | Swollen hepatocytes + inflammation narrow sinusoids acutely |
| Amyloidosis | Amyloid deposits compress sinusoids |
| Infiltrative malignancy | Cancer cells fill and compress sinusoids |
| Vitamin A toxicity | Causes hepatic stellate cell activation and sinusoidal fibrosis |
C. Post-Sinusoidal (after sinusoids, before hepatic veins)
| Cause | Notes |
|---|---|
| Veno-occlusive disease (VOD) / Sinusoidal Obstruction Syndrome (SOS) | Fibrous obliteration of terminal hepatic venules - seen after bone marrow transplant, chemotherapy (oxaliplatin), or pyrrolizidine alkaloid toxicity (bush tea) |
3. POST-HEPATIC Causes
(Block after blood leaves the liver)
Liver is often normal initially. Pressure backs up from the heart/hepatic veins into the liver.
| Cause | Notes |
|---|---|
| Budd-Chiari Syndrome | Thrombosis of hepatic veins - classic triad: abdominal pain, ascites, hepatomegaly. Associated with hypercoagulable states (polycythemia vera, JAK2 mutation, OCP use, pregnancy) |
| Inferior vena cava obstruction | Thrombosis or membrane/web blocking IVC |
| Severe right heart failure | Right ventricle cannot accept blood → backs up into hepatic veins |
| Constrictive pericarditis | Stiff pericardium prevents right heart from filling → same result |
| Restrictive cardiomyopathy | Same mechanism as above |
| Tricuspid regurgitation | Severe TR transmits back-pressure into hepatic veins |
Key feature: These patients develop "cardiac cirrhosis" over time - the liver is congested but the primary problem is cardiac/vascular.
Quick Memory Table
| Level | Common Causes | Key Feature |
|---|---|---|
| Pre-hepatic | Portal vein thrombosis, splenomegaly | Normal liver function, HVPG normal |
| Pre-sinusoidal (intrahepatic) | Schistosomiasis, PBC, sarcoid | Portal fibrosis, lobular architecture preserved |
| Sinusoidal (intrahepatic) | Cirrhosis (most common) | Elevated HVPG, liver failure features |
| Post-sinusoidal (intrahepatic) | Veno-occlusive disease | History of BMT/chemo |
| Post-hepatic | Budd-Chiari, right heart failure | Hepatomegaly, ascites, cardiac signs |
Worldwide vs. Western World
| Region | Most Common Cause |
|---|---|
| Western countries | Cirrhosis (alcohol #1, then NAFLD, viral hepatitis) |
| Worldwide (global) | Schistosomiasis (hundreds of millions infected) |
| Children | Portal vein thrombosis (often from neonatal umbilical vein catheterization) |
| Young women with clots | Budd-Chiari (OCP, pregnancy, thrombophilia) |
Simple Way to Remember
PRE-hepatic → Something BLOCKS the portal vein before it enters the liver
(clot, compression, too much flow)
INTRA-hepatic → The liver itself is DAMAGED or INFILTRATED
(scarred = cirrhosis / granulomas = schistosomiasis)
POST-hepatic → Blood cannot LEAVE the liver
(hepatic veins blocked / heart too weak to pull blood forward)
All three ultimately cause the same result: blood dams up in the portal system → pressure rises → varices, ascites, splenomegaly.
This is a shared conversation. Sign in to Orris to start your own chat.