Here is your complete, topic-wise viva preparation guide. Each answer is written in the concise, direct style examiners expect, followed by likely cross-questions.
MBBS 2nd Year Pharmacology - Complete Viva Preparation
SECTION 1: GENERAL PHARMACOLOGY
PHARMACOKINETICS
Q: What is pharmacokinetics?
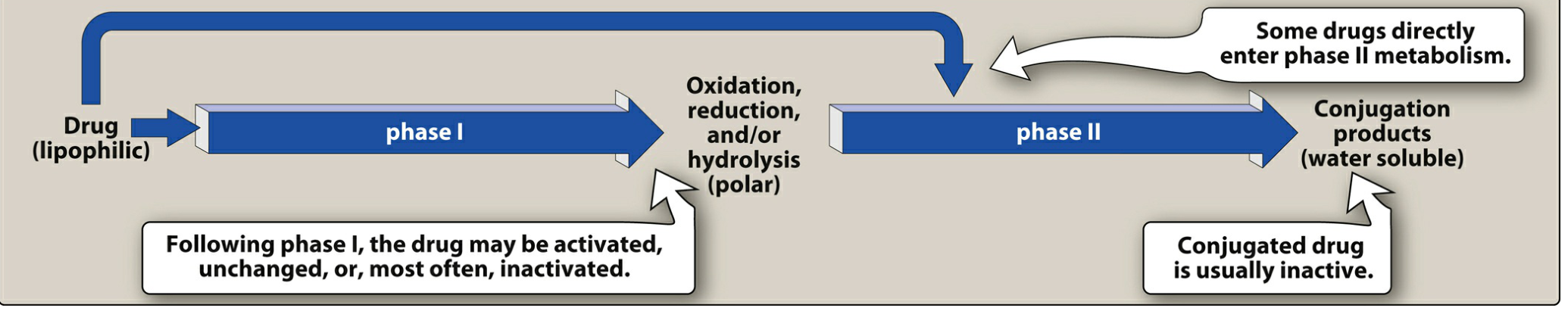
A: Pharmacokinetics is "what the body does to the drug." It involves four processes - Absorption, Distribution, Metabolism, and Excretion (ADME).
Q: Define drug absorption.
A: Absorption is the movement of a drug from its site of administration into the systemic circulation.
Q: What are the factors affecting drug absorption?
Drug-related factors:
- Physicochemical properties: lipid solubility, molecular weight, ionization (pKa)
- Drug formulation: tablet vs liquid, coating, particle size
- Route of administration
Patient-related factors:
- GI motility (faster motility = less absorption)
- Gastric pH
- Gut surface area (reduced in bowel disease)
- Presence of food in stomach
- Blood flow to absorption site
- Age (infants/elderly have altered GI function)
Cross Q: How does food affect drug absorption?
Food generally delays absorption (slows gastric emptying). Some drugs need food (e.g., griseofulvin - fat-soluble); others are better on empty stomach (e.g., ampicillin).
Q: Effect of pH on drug absorption?
A: Follows Henderson-Hasselbalch principle - non-ionized form is lipid-soluble and better absorbed.
- Weak acids (e.g., aspirin, pKa 3.5): absorbed better in acidic stomach (non-ionized here)
- Weak bases (e.g., morphine): absorbed better in alkaline small intestine (non-ionized here)
Key Rule: Weak acids absorbed in stomach; weak bases absorbed in intestine.
Q: Examples of different processes of absorption?
| Process | Example |
|---|
| Passive diffusion | Most drugs (aspirin, barbiturates) |
| Active transport | Levodopa, methyldopa, 5-FU |
| Facilitated diffusion | Vitamin B12 |
| Pinocytosis | Proteins, vaccines |
| Filtration | Water, small ions |
Q: Define drug distribution.
A: Distribution is the reversible transfer of drug from systemic circulation to tissues and organs.
Q: Factors affecting distribution?
- Lipid solubility (higher = wider distribution)
- Plasma protein binding
- Tissue binding
- Blood-brain barrier (lipid-soluble drugs cross)
- Placental barrier
- Volume of distribution (Vd)
Q: What is plasma protein binding? Difference between protein-bound and free drug?
| Feature | Protein-Bound Drug | Free Drug |
|---|
| Pharmacologically active? | No | Yes (active) |
| Filtered at glomerulus? | No | Yes |
| Metabolized? | No | Yes |
| Crosses membranes? | No | Yes |
| Acts as reservoir? | Yes | No |
Cross Q: Which plasma protein do acidic drugs bind to? Albumin. Basics drugs? Alpha-1 acid glycoprotein.
Cross Q: Name two highly protein-bound drugs. Warfarin (99%), Diazepam (98%).
Cross Q: What is drug displacement interaction? When two drugs compete for same binding site, the displaced drug has increased free fraction - risk of toxicity (e.g., warfarin + aspirin).
Q: Define drug excretion.
A: Excretion is the irreversible removal of the drug or its metabolites from the body.
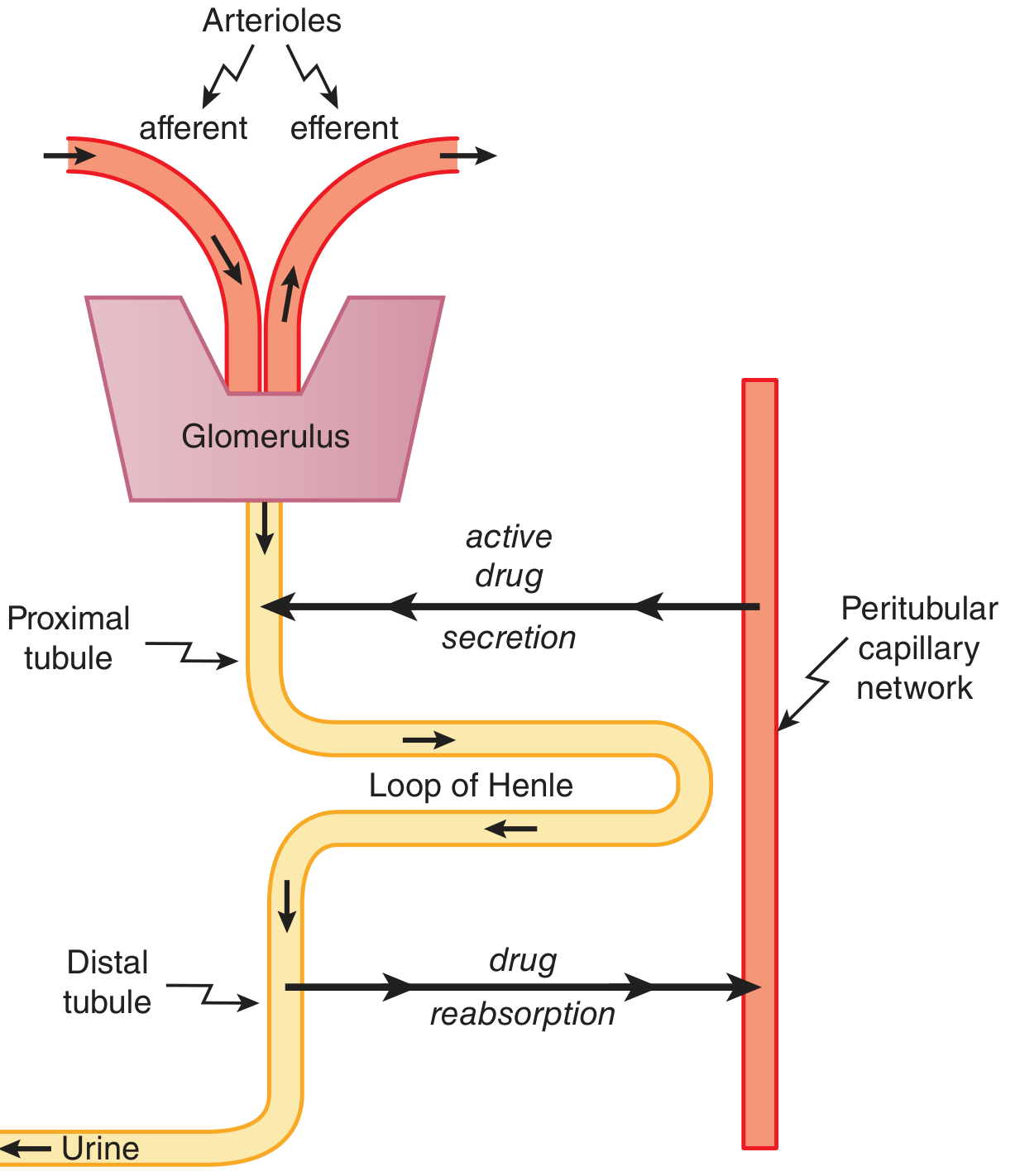
Q: What is tubular secretion?
A: Active, carrier-mediated transport of drugs from peritubular capillaries into the proximal tubular lumen. Involves two systems:
- Organic anion transporters (OAT1, OAT3) - for acidic drugs (penicillin, furosemide, methotrexate)
- Organic cation transporters (OCT) - for basic drugs (dopamine, histamine)
Cross Q: Give a clinical example of tubular secretion interaction. Probenecid competes with penicillin at OAT, blocking its secretion - prolongs penicillin's action.
Q: Role of urine pH in drug excretion?
(Covered in detail in your previous session)
- Alkaline urine: increases excretion of weak acids (salicylate, phenobarbital) - used in aspirin overdose
- Acidic urine: increases excretion of weak bases (amphetamine) - rarely used clinically due to risk of rhabdomyolysis
Q: Substances excreted through feces?
- Unabsorbed orally ingested drugs
- Drugs excreted in bile (enterohepatic circulation - digoxin, rifampicin, oral contraceptives)
- Drugs secreted directly into intestine
Q: Substances excreted through saliva?
- Heavy metals (lead, mercury, bismuth)
- Metronidazole (causes metallic taste)
- Urea (in renal failure - uremic frost equivalent in saliva)
- Iodides, bromides
RECEPTORS
Q: Define receptor.
A: A receptor is a macromolecule (usually a protein) that specifically binds a drug or endogenous ligand and initiates a pharmacological response. It has two domains: ligand-binding domain and effector domain.
Q: Regulation of receptors?
- Up-regulation (supersensitivity): Chronic antagonist use or denervation increases receptor number (e.g., beta-blockers increase beta-receptor density)
- Down-regulation (desensitization): Chronic agonist use decreases receptor number (e.g., chronic beta-agonist use)
Cross Q: Clinical example of up-regulation? Sudden stopping of beta-blockers causes rebound tachycardia due to supersensitive beta-receptors.
Q: Types of neurotransmitters?
| Type | Examples |
|---|
| Cholinergic | Acetylcholine |
| Adrenergic | Norepinephrine, Epinephrine |
| Amino acid | GABA, Glutamate, Glycine |
| Peptide | Substance P, Enkephalins, Endorphins |
| Monoamine | Dopamine, Serotonin, Histamine |
Q: Types of adrenergic receptors and their sites?
| Receptor | Location | Response |
|---|
| α1 | Blood vessels, eye, bladder neck | Vasoconstriction, mydriasis, urinary retention |
| α2 | Presynaptic nerve terminals, CNS | Inhibits NE release; CNS: reduces BP |
| β1 | Heart, kidney (JGA) | ↑ HR, ↑ contractility; renin release |
| β2 | Bronchi, blood vessels, uterus, liver | Bronchodilation, vasodilation, uterine relaxation |
| β3 | Adipose tissue | Lipolysis |
Q: Cholinergic receptors and muscarinic receptors?
Cholinergic receptors:
- Nicotinic (NM): Neuromuscular junction → muscle contraction
- Nicotinic (NN): Autonomic ganglia, adrenal medulla
- Muscarinic (M1-M5): Post-ganglionic parasympathetic synapses
Muscarinic receptor subtypes:
| Subtype | Location | Effect |
|---|
| M1 | Gastric glands, CNS, ganglia | ↑ gastric acid, CNS excitation |
| M2 | Heart (SA node, AV node) | ↓ HR, ↓ conduction |
| M3 | Smooth muscle, glands, eye | Contraction, secretion, miosis, lacrimation |
Memory: M2 = heart (2 chambers pumping), M3 = secretion, smooth muscle.
Q: Define agonist and partial agonist. Examples?
| Term | Definition | Example |
|---|
| Agonist (full) | Binds receptor + produces maximal response (efficacy = 1) | Morphine, adrenaline, salbutamol |
| Partial agonist | Binds receptor but produces submaximal response even at full occupancy (efficacy <1) | Buprenorphine, aripiprazole, pindolol |
| Antagonist | Binds but produces no response (efficacy = 0) | Naloxone, atropine, propranolol |
Cross Q: What happens if a partial agonist is given with a full agonist? The partial agonist acts as an antagonist by competing for receptors while producing less effect - NET EFFECT: reduced response (e.g., buprenorphine reduces morphine's effect).
Q: Non-receptor mediated drug mechanisms with examples?
| Mechanism | Example |
|---|
| Physical action (osmosis) | Mannitol (osmotic diuretic), antacids |
| Chemical neutralization | Antacids (NaHCO₃ + HCl → NaCl + H₂O) |
| Metal chelation | Dimercaprol, EDTA in heavy metal poisoning |
| Enzyme inhibition (non-receptor) | Acetazolamide (inhibits carbonic anhydrase) |
| Membrane stabilization | Local anesthetics (non-specific Na⁺ channel block) |
| DNA intercalation | Chloroquine, doxorubicin |
DRUG DEVELOPMENT & PHARMACOGENETICS
Q: Steps in drug development?
- Drug discovery - natural sources, synthesis, screening
- Preclinical testing - in vitro, animal studies (safety, toxicity)
- Phase I trials - healthy volunteers, safety, dose-finding, pharmacokinetics (20-100 subjects)
- Phase II trials - patients, efficacy and safety (100-300)
- Phase III trials - large multicenter RCTs (1000-3000 patients), comparison with placebo/standard
- Regulatory approval (CDSCO in India, FDA in USA)
- Phase IV (post-marketing surveillance) - long-term safety, rare ADRs
Cross Q: What is Phase 0? Microdosing studies in humans - sub-therapeutic doses to study PK/PD.
Q: Recombinant DNA technology with examples?
A: Genetic engineering technique that inserts human DNA coding for a protein into a vector (plasmid/virus) → expressed in bacteria/yeast/CHO cells → purified protein used as drug.
Examples:
- Human insulin (produced in E. coli) - replaced animal insulin
- Human growth hormone (somatropin)
- Erythropoietin (EPO) - for anemia in CKD
- Tissue plasminogen activator (tPA/alteplase) - thrombolysis
- Interferon-alpha, beta, gamma
- Hepatitis B vaccine (recombinant HBsAg)
- Monoclonal antibodies (trastuzumab, rituximab)
Q: Succinylcholine and pharmacogenetics/pharmacogenomics?
A: Succinylcholine is normally hydrolyzed by plasma pseudocholinesterase (butyrylcholinesterase) within 5-10 minutes. Some individuals have a genetic variant of pseudocholinesterase with reduced dibucaine number (normal = 80, atypical = 20).
- In these patients, succinylcholine is NOT hydrolyzed normally
- Results in prolonged neuromuscular blockade - "scoline apnea" lasting 2-3 hours
- Management: mechanical ventilation until drug wears off; fresh frozen plasma (contains pseudocholinesterase)
This is the classic pharmacogenetics example in MBBS viva - always mention dibucaine number.
DOSAGE & ROUTES
Q: Types of doses?
| Type | Definition | Example |
|---|
| Therapeutic dose | Dose producing desired effect without toxicity | Paracetamol 500 mg |
| Minimum effective dose | Smallest dose producing desired effect | - |
| Maximum tolerated dose | Largest dose without toxic effects | - |
| Lethal dose (LD50) | Dose killing 50% of experimental animals | - |
| Loading dose | Initial large dose to rapidly achieve therapeutic level | Digoxin, amiodarone |
| Maintenance dose | Dose to maintain steady-state plasma level | Daily digoxin |
| Ceiling dose | Dose beyond which no additional effect occurs | NSAIDs, diuretics |
Q: Four parenteral routes with examples?
| Route | Example Drugs | Key Points |
|---|
| Intravenous (IV) | Morphine, diazepam, heparin | Fastest onset; 100% bioavailability; risk of embolism |
| Intramuscular (IM) | Penicillin G, vaccines, diclofenac | Depot preparations possible |
| Subcutaneous (SC) | Insulin, heparin, adrenaline | Slow absorption, suitable for self-injection |
| Intrathecal | Morphine, bupivacaine, methotrexate | Bypasses blood-brain barrier |
Others: intra-arterial (streptokinase), intra-articular (steroids), epidural.
Q: Advantages and disadvantages of rectal route?
| Advantages | Disadvantages |
|---|
| Avoids first-pass metabolism (50%) | Irregular, unpredictable absorption |
| Useful when oral route unavailable (vomiting, unconscious) | Patient non-compliance, discomfort |
| Useful in pediatrics | Cannot use if anorectal disease |
| Local action possible (enemas) | Irritation of rectal mucosa |
Q: Solid dosage forms?
- Tablets - compressed powder; most common
- Capsules - gelatin shell; mask unpleasant taste
- Enteric-coated tablets - coating resists gastric acid (aspirin EC, diclofenac EC)
- Sustained-release (SR/XL/ER) - slow release over time; reduces dosing frequency
- Lozenges - dissolve in mouth (for local throat action)
- Suppositories - rectal/vaginal solid form
Q: WHO definition of ADR?
A: "A response to a drug which is noxious and unintended, and which occurs at doses normally used in man for prophylaxis, diagnosis, or therapy of disease, or for modification of physiological function."
Key phrase: "noxious and unintended" + "at normal doses" (excludes overdose/poisoning).
SECTION 2: AUTONOMIC NERVOUS SYSTEM
CHOLINERGIC DRUGS
Q: Classification of indirect cholinergic drugs?
Indirect cholinergic drugs work by inhibiting acetylcholinesterase (anticholinesterase agents):
Reversible:
- Short-acting: Edrophonium (diagnosis of myasthenia)
- Medium-acting: Neostigmine, Pyridostigmine (quaternary ammonium - does NOT cross BBB)
- Lipid-soluble (crosses BBB): Physostigmine, Donepezil, Rivastigmine, Galantamine
Irreversible:
- Organophosphates: Echothiophate (glaucoma), Parathion, Malathion (insecticides), Nerve gases (Sarin, VX)
Q: Neostigmine - mechanism?
A: Neostigmine is a reversible, competitive inhibitor of acetylcholinesterase. It binds the anionic and esteratic sites of AChE, preventing breakdown of acetylcholine. This accumulates ACh at all cholinergic synapses (NMJ, autonomic ganglia, parasympathetic effectors).
Uses: Myasthenia gravis, reversal of non-depolarizing NMB, postoperative urinary retention/paralytic ileus.
Does NOT cross BBB (quaternary ammonium compound).
Q: Non-depolarizing neuromuscular blockers (NDNMBs)?
- Short-acting: Mivacurium
- Intermediate: Vecuronium, Rocuronium, Atracurium, Cisatracurium
- Long-acting: Pancuronium, d-Tubocurarine
Mechanism: Competitive antagonism at nicotinic NM receptors - block ACh binding without depolarizing end-plate.
Reversal: Neostigmine + atropine (to block muscarinic side effects of neostigmine); OR Sugammadex (for rocuronium/vecuronium - forms inclusion complex).
Q: Succinylcholine (Suxamethonium)?
- Class: Depolarizing neuromuscular blocker
- Mechanism: Acts as ACh mimic at NMJ → persistent depolarization → initial fasciculations → flaccid paralysis (receptor cannot repolarize)
- Duration: Ultra-short (5-10 min) - hydrolyzed by plasma pseudocholinesterase
- Uses: Rapid sequence intubation (RSI), short procedures
- Side effects: Hyperkalemia (contraindicated in burns, crush injury, denervation), malignant hyperthermia (with halothane), raised IOP, bradycardia, prolonged block (genetic - see pharmacogenetics above)
- NOT reversed by neostigmine (no competitive antagonism)
Q: Differences between depolarizing and non-depolarizing NMBs?
| Feature | Depolarizing (Succinylcholine) | Non-depolarizing (Vecuronium) |
|---|
| Mechanism | ACh mimic - persistent depolarization | Competitive antagonist |
| Initial fasciculations | Yes | No |
| Reversal | Not reversible (wait for metabolism) | Neostigmine or Sugammadex |
| Effect of AChE inhibitors | Potentiated (more block) | Antagonized (reversed) |
| Tetanic stimulation | Sustained response | Fade |
| Post-tetanic facilitation | No | Yes |
| Duration | Ultra-short | Variable (short to long) |
ORGANOPHOSPHATE (OPC) POISONING
Q: Mechanism of OPC poisoning?
A: Organophosphates irreversibly phosphorylate the esteratic site of acetylcholinesterase → AChE permanently inactivated → ACh accumulates at all cholinergic synapses.
Features (SLUDGE + Nicotinic + CNS):
- Muscarinic (SLUDGE): Salivation, Lacrimation, Urination, Defecation, GI cramps, Emesis + Bradycardia, Bronchoconstriction, Miosis
- Nicotinic: Muscle fasciculations → weakness → paralysis; hypertension, tachycardia
- CNS: Anxiety, seizures, coma, respiratory failure
Mnemonic for muscarinic effects: DUMBELS - Diarrhea, Urination, Miosis, Bradycardia, Emesis, Lacrimation, Salivation
Q: Antidote, dose, and mechanism for OPC poisoning?
Atropine:
- Antidote for muscarinic effects only
- Dose: 2-4 mg IV every 5-10 min until muscarinic features dry up (secretions dry, bronchospasm resolves) - no upper limit; massive doses may be needed
- Mechanism: Competitive muscarinic antagonist - blocks ACh at muscarinic receptors
Pralidoxime (2-PAM):
- Antidote for both muscarinic AND nicotinic effects
- Dose: 1-2 g IV over 15-30 min, then infusion
- Mechanism: Reactivates acetylcholinesterase by cleaving the phosphate-AChE bond (if given BEFORE "aging" - irreversible conformational change, usually within 24-48 hours)
- Must be given early - becomes ineffective after aging
Cross Q: Which effects does atropine NOT treat in OPC? Nicotinic effects (muscle fasciculations, paralysis) - only pralidoxime helps these.
Q: Complete management of OPC poisoning?
- Remove from exposure - decontamination, remove clothing, wash skin
- ABC - airway (intubation if needed), suction secretions
- Atropine - large doses IV until secretions dry
- Pralidoxime - early administration
- Benzodiazepines (diazepam) - for seizures
- Avoid: succinylcholine (potentiated), organophosphate-class insecticide exposure
- Supportive care - mechanical ventilation, fluids
ATROPINE
Q: Signs of atropine poisoning ("Hot as a hare, Dry as a bone, Red as a beet, Mad as a hatter, Blind as a bat")?
- Hyperthermia (↓ sweating) - "Hot as a hare"
- Dry skin and mouth - "Dry as a bone"
- Flushing (vasodilation) - "Red as a beet"
- Delirium, hallucinations - "Mad as a hatter"
- Mydriasis, cycloplegia (blurred vision) - "Blind as a bat"
- Also: Tachycardia, urinary retention, constipation
Treatment: Physostigmine (crosses BBB - reverses CNS effects too)
Q: Atropine-like drugs (antimuscarinics)?
| Drug | Special Feature |
|---|
| Hyoscine (scopolamine) | Better for motion sickness; CNS depression (unlike atropine which causes excitation) |
| Ipratropium | Inhaled; bronchodilation in COPD/asthma; no systemic effects |
| Tiotropium | Long-acting; once-daily; COPD |
| Oxybutynin, Solifenacin | Overactive bladder |
| Benztropine, Trihexyphenidyl | Parkinson's disease |
| Dicyclomine | Irritable bowel syndrome |
| Homatropine, Tropicamide | Mydriatic/cycloplegic (eye) |
Q: Why is atropine used as pre-anaesthetic medication?
A: To reduce secretions (anti-sialagogue effect) caused by inhalational anesthetics and airway manipulation. Additional benefits:
- Prevents bradycardia from vagal stimulation during intubation/surgery
- Prevents bronchospasm
- Reduces gastric acid secretion (slight)
- Causes sedation with hyoscine (if hyoscine used instead)
Modern anesthetics are less irritating, so atropine as pre-med is now selective, not routine.
Q: Ophthalmic use of atropine?
- Cycloplegia (ciliary muscle paralysis) - refraction testing in children
- Mydriasis - fundus examination, pre/post intraocular surgery
- Treatment of uveitis/iritis - prevents synechiae formation
- Amblyopia treatment - penalizing the better eye
Q: Mydriatic drugs. Which is preferred in elderly and why?
Mydriatics:
| Drug | Mechanism | Duration | Cycloplegia? |
|---|
| Atropine | Muscarinic block | 7-10 days | Yes |
| Homatropine | Muscarinic block | 1-3 days | Yes (mild) |
| Tropicamide | Muscarinic block | 4-6 hours | Mild |
| Cyclopentolate | Muscarinic block | 24 hours | Yes |
| Phenylephrine | α1 agonist (dilates dilator pupillae) | 4-6 hours | No |
Preferred in elderly: Tropicamide (short-acting) or Phenylephrine
Why? Elderly patients are at high risk of acute angle-closure glaucoma with long-acting mydriatics (atropine, homatropine), because:
- Elderly have shallow anterior chambers (lens thickens with age)
- Pupil dilation obstructs trabecular drainage → ↑ IOP
- Tropicamide wears off quickly (4-6 hrs), reducing this risk
SECTION 3: ADRENERGIC DRUGS
Q: Classification of indirect adrenergic agonists?
Indirect adrenergic agonists work by increasing norepinephrine at the synapse:
- Releasing agents: Amphetamine, tyramine, ephedrine
- Uptake inhibitors: Cocaine, tricyclic antidepressants (block reuptake of NE)
- MAO inhibitors: Phenelzine, tranylcypromine (block degradation of NE)
Q: Beta agonists - classification and examples?
| Selectivity | Drug | Use |
|---|
| Non-selective β1+β2 | Isoprenaline | Bradycardia (historical) |
| Selective β1 | Dobutamine | Acute heart failure (inotrope) |
| Selective β2 | Salbutamol, Terbutaline | Asthma bronchodilation |
| Long-acting β2 | Salmeterol, Formoterol | Maintenance asthma/COPD |
Q: Why is adrenaline used in anaphylaxis and shock?
A: Adrenaline (epinephrine) acts on both alpha and beta receptors:
- α1 effects: Vasoconstriction → ↑ BP, reduces angioedema and urticaria
- β1 effects: ↑ HR and contractility → ↑ cardiac output
- β2 effects: Bronchodilation → relieves bronchospasm; inhibits mast cell mediator release
- It is the only drug that addresses ALL components of anaphylaxis simultaneously.
Cross Q: Route in anaphylaxis? IM into anterolateral thigh (NOT IV unless cardiac arrest).
Q: Beta blocker classification?
| Generation | Selectivity | Examples |
|---|
| 1st gen (non-selective) | β1 + β2 | Propranolol, Timolol, Nadolol |
| 2nd gen (cardioselective) | β1 selective | Atenolol, Metoprolol, Bisoprolol |
| 3rd gen (vasodilating) | β1 + α1 block OR β3/NO | Carvedilol (α+β), Labetalol (α+β), Nebivolol (β1 + ↑NO) |
Q: Atenolol vs Propranolol?
| Feature | Atenolol | Propranolol |
|---|
| Receptor selectivity | β1 selective | Non-selective (β1 + β2) |
| Lipid solubility | Low (hydrophilic) | High (lipophilic) |
| CNS penetration | Poor | Good (nightmares, depression) |
| Route | Oral (poor IV use) | Oral + IV |
| Renal excretion | Mainly renal | Mainly hepatic |
| Use in asthma | Safer (relative) | Contraindicated |
| First-pass effect | Minimal | Extensive (large first-pass) |
SECTION 4: ANTIHYPERTENSIVE DRUGS
Q: Drugs used in hypertension with diabetes?
- First-line: ACE inhibitors or ARBs (nephroprotective - reduce proteinuria, slow diabetic nephropathy)
- If still not controlled: Add thiazide or CCB (amlodipine)
- Avoid: Beta-blockers (mask hypoglycemia symptoms, worsen insulin resistance)
Q: Why are ACE inhibitors first-line in hypertension with DM?
- Reduce blood pressure
- Reduce intraglomerular pressure (dilate efferent arteriole) → reduce proteinuria
- Slow progression of diabetic nephropathy
- Cardioprotective
- Reduce microalbuminuria even before frank proteinuria
Q: ACE inhibitor vs ARB comparison?
| Feature | ACE Inhibitor | ARB |
|---|
| Mechanism | Blocks conversion of Ang I → Ang II | Blocks AT1 receptor |
| Cough | Yes (↑ bradykinin) | No |
| Angioedema | More common | Less common but possible |
| Effect on bradykinin | ↑ bradykinin (beneficial for BP) | No effect |
| Use if cough | Switch to ARB | First choice |
| Examples | Enalapril, Ramipril, Lisinopril | Losartan, Valsartan, Telmisartan |
Cross Q: Why do ACE inhibitors cause cough? ACE also breaks down bradykinin. When ACE is inhibited, bradykinin accumulates → irritates bronchi → dry, persistent cough in ~10-15% patients.
Q: Why ACE inhibitors are NOT used in bronchial asthma?
- Bradykinin accumulation → airway inflammation, bronchoconstriction → cough and potential bronchospasm
- Use ARBs instead (do not affect bradykinin)
Q: Hypertension with bronchial asthma?
- Use: CCB (amlodipine), ARBs
- Avoid: Beta-blockers (β2 block → bronchospasm), ACE inhibitors (cough/bronchospasm)
Q: Hypertension with obesity and nephropathy?
- ACE inhibitor or ARB (nephroprotection)
- Add CCB or thiazide if needed
Q: Hypertension with diabetic neuropathy?
- Duloxetine or pregabalin (for neuropathic pain)
- Antihypertensive: ACE inhibitor/ARB
Q: Prazosin - mechanism, adverse effects, first-dose phenomenon?
- Mechanism: Selective α1 antagonist → vasodilation (arteriolar + venous) → ↓ BP
- Adverse effects: First-dose phenomenon, postural hypotension, reflex tachycardia, nasal stuffiness, headache, fluid retention
- First-dose phenomenon: Sudden severe hypotension and syncope after the first dose due to loss of α1-mediated vascular tone without reflex compensation
- Prevention: Start with very low dose (0.5 mg) at bedtime, with the patient lying down; titrate gradually
Q: Methyldopa - mechanism, adverse effects, use in PIH?
- Mechanism: Centrally acting antihypertensive. Converted to alpha-methylnorepinephrine in CNS → stimulates α2 receptors (presynaptic) → reduces sympathetic outflow → ↓ BP
- Adverse effects: Sedation (most common), dry mouth, positive Coombs test (hemolytic anemia - rare), lupus-like syndrome, hepatotoxicity
- Drug of choice in pregnancy-induced hypertension (PIH): Safe for fetus; crosses placenta without teratogenicity; long safety track record
Cross Q: What is the drug of choice for severe/acute PIH? Labetalol or Hydralazine IV. For maintenance: Methyldopa, Nifedipine.
Q: Hydralazine - mechanism?
- Direct vasodilator → relaxes arteriolar smooth muscle (mainly) → ↓ peripheral resistance
- Mechanism: Increases cGMP in vascular smooth muscle; may open K⁺ channels
- Reflex effects: Tachycardia, fluid retention, ↑ renin release
- Uses: PIH (IV), heart failure with nitrates (combination), hypertensive crisis
- Side effect: Lupus-like syndrome (dose-dependent, in slow acetylators)
Q: Thiazide diuretics - mechanism, hyperuricemia, adverse effects?
- Mechanism: Inhibit Na⁺/Cl⁻ cotransporter in distal convoluted tubule (DCT) → ↑ Na⁺ and water excretion → ↓ plasma volume → ↓ BP
- Why hyperuricemia? Thiazides compete with uric acid for proximal tubule secretion via OAT transporters. Also volume contraction → ↑ urate reabsorption → hyperuricemia → can precipitate gout
- Adverse effects: Hypokalemia, hyponatremia, hyperuricemia, hyperglycemia, hyperlipidemia, hypercalcemia (remember: thiazides retain calcium - used in hypercalciuria/nephrolithiasis), erectile dysfunction
SECTION 5: DIURETICS
Q: Diuretics acting on DCT and collecting duct?
DCT (Distal Convoluted Tubule):
- Thiazides (hydrochlorothiazide, chlorthalidone) - block Na/Cl cotransporter
Collecting Duct:
- Potassium-sparing diuretics:
- Aldosterone antagonists: Spironolactone, Eplerenone (block mineralocorticoid receptor → ↓ Na⁺ reabsorption, ↓ K⁺ secretion)
- Na⁺ channel blockers: Amiloride, Triamterene (block epithelial Na⁺ channel - ENaC)
Q: Furosemide vs Thiazide?
| Feature | Furosemide | Thiazide |
|---|
| Site of action | Loop of Henle (thick ascending) | DCT |
| Transporter blocked | NKCC2 (Na-K-2Cl) | NCC (Na-Cl) |
| Potency | High (ceiling diuretic) | Moderate |
| Calcium excretion | Increases Ca²⁺ excretion | Decreases Ca²⁺ excretion |
| Glucose effect | Can cause hyperglycemia | More pronounced hyperglycemia |
| Use in renal failure | Effective (GFR <30) | Ineffective if GFR <30 |
| Use in hypercalcemia | Yes (↑Ca excretion) | No |
| Use in nephrolithiasis | No | Yes (↓Ca excretion) |
Q: Furosemide vs Spironolactone?
| Feature | Furosemide | Spironolactone |
|---|
| Site | Loop of Henle | Collecting duct |
| Potassium effect | Hypokalemia | Hyperkalemia (K-sparing) |
| Mechanism | NKCC2 block | Aldosterone antagonist |
| Use in cirrhotic ascites | Yes (with spironolactone) | Yes (preferred as aldosterone is elevated) |
| Anti-androgen effect | No | Yes (gynecomastia, menstrual irregularity) |
Classic combination: Furosemide + Spironolactone in hepatic ascites (ratio 40:100 mg to maintain normokalemia).
Q: Drug for leg swelling (edema)?
- Cardiac edema: Furosemide ± spironolactone
- Cirrhotic ascites/edema: Spironolactone (preferred) + furosemide
- Hypertensive: Thiazide
- Nephrotic: Furosemide
SECTION 6: DIABETES MELLITUS
Q: Euglycemic agents (drugs that don't cause hypoglycemia) with examples?
- Metformin (biguanide)
- Acarbose (alpha-glucosidase inhibitor)
- GLP-1 agonists (exenatide, liraglutide) - very low risk of hypoglycemia alone
- DPP-4 inhibitors (sitagliptin, linagliptin) - weight neutral, low hypoglycemia risk
- SGLT-2 inhibitors (empagliflozin, dapagliflozin)
- Thiazolidinediones (pioglitazone) - very low hypoglycemia risk
Q: Drugs used for diabetes in pregnancy?
- Insulin is the drug of choice (does not cross placenta; safest)
- Metformin is used in some countries (some evidence for safety, used for GDM)
- Sulfonylureas, GLP-1 agonists, SGLT-2 inhibitors - AVOID (teratogenic potential or insufficient data)
Q: Insulin preparations - classification by duration?
| Type | Onset | Peak | Duration | Examples |
|---|
| Ultra-rapid | 10-15 min | 1 hr | 2-4 hrs | Lispro, Aspart, Glulisine |
| Short-acting (Regular) | 30 min | 2-3 hrs | 6-8 hrs | Regular/Soluble insulin |
| Intermediate | 1-2 hrs | 4-8 hrs | 12-18 hrs | NPH (Isophane) |
| Long-acting | 1-2 hrs | Peakless | 20-24 hrs | Glargine, Detemir |
| Ultra-long | 6 hrs | Peakless | 42+ hrs | Degludec |
Q: Mechanism of insulin?
- Binds insulin receptor (tyrosine kinase receptor) on cell membrane
- Autophosphorylation → activates IRS proteins
- PI3K pathway → GLUT-4 translocation to membrane → ↑ glucose uptake
- Promotes glycogen synthesis (liver, muscle), protein synthesis, fat storage
- Inhibits gluconeogenesis, glycogenolysis, lipolysis
Q: Adverse effects of insulin?
- Hypoglycemia (most common and dangerous)
- Lipodystrophy at injection site (lipoatrophy or lipohypertrophy)
- Weight gain
- Hypokalemia (insulin shifts K⁺ into cells)
- Edema (sodium retention)
- Insulin resistance (rare)
Cross Q: Why is insulin given in trauma/accident in diabetic patients? Trauma → stress response → counter-regulatory hormones (cortisol, glucagon, adrenaline) → hyperglycemia. Insulin is given to control blood glucose and prevent DKA. Also, insulin has anabolic effects promoting wound healing.
Q: Metformin - indications, mechanism, adverse effects?
Mechanism:
- Primary: Activates AMPK → inhibits hepatic gluconeogenesis (main effect)
- Improves peripheral insulin sensitivity
- Delays GI glucose absorption
- Does NOT stimulate insulin secretion → no hypoglycemia
Indications:
- First-line in Type 2 DM (especially obese patients)
- PCOS (improves insulin resistance)
- Pre-diabetes prevention
Adverse effects:
- GI: nausea, diarrhea, abdominal pain (most common - take with food)
- Lactic acidosis (rare but serious - accumulation in hypoxic states)
- Vitamin B12 deficiency (long-term use)
- Metallic taste
Contraindications: eGFR <30, acute heart failure, acute liver failure, severe infection, iodinated contrast dye (hold for 48 hrs), surgery
Q: GLP-1 agonists mechanism?
- Mimic glucagon-like peptide-1 (GLP-1) - incretin hormone
- Mechanism:
- Stimulate insulin secretion in a glucose-dependent manner (only when glucose is high → no hypoglycemia)
- Suppress glucagon secretion
- Delay gastric emptying → reduce postprandial glucose
- Central satiety → weight loss
- Examples: Exenatide, Liraglutide, Semaglutide
- Benefits: Weight loss, cardiovascular protection (liraglutide, semaglutide), nephroprotection
Q: Linagliptin mechanism?
- DPP-4 inhibitor (gliptin class)
- Dipeptidyl peptidase-4 is the enzyme that degrades GLP-1 and GIP (incretin hormones)
- Linagliptin blocks DPP-4 → GLP-1 levels increase → ↑ glucose-dependent insulin secretion + ↓ glucagon
- Advantage over other gliptins: Excreted mainly via bile (not kidney) → safe in renal impairment without dose adjustment
SECTION 7: ANTIANGINAL DRUGS
Q: Classification of antianginal drugs?
- Nitrates: GTN (sublingual, patch), Isosorbide mononitrate/dinitrate
- Beta-blockers: Propranolol, Atenolol, Metoprolol
- Calcium channel blockers:
- Non-dihydropyridines (heart rate-lowering): Verapamil, Diltiazem
- Dihydropyridines (vasodilating): Amlodipine, Nifedipine
- Other: Ranolazine (late Na⁺ channel blocker), Ivabradine (If channel blocker)
Q: Calcium channel blockers - effect on hypertension and angina?
Hypertension:
- Dihydropyridines (amlodipine, nifedipine): Vasodilate arterioles → ↓ peripheral resistance → ↓ BP
- Verapamil/Diltiazem: Also lower BP but mainly via cardiac effects
Angina:
- Dilate coronary arteries → ↑ oxygen supply
- Reduce afterload → ↓ cardiac work → ↓ oxygen demand
- Verapamil/Diltiazem: Also ↓ HR → ↑ diastolic filling time → ↑ coronary perfusion
Cross Q: Which CCB is preferred in angina with tachycardia? Verapamil or Diltiazem (rate-limiting CCBs). NOT amlodipine (causes reflex tachycardia).
Q: GTN (glyceryl trinitrate) adverse effects?
- Headache (most common - vasodilation of meningeal vessels)
- Flushing, postural hypotension, syncope
- Tolerance (tachyphylaxis) - with continuous use; prevent by nitrate-free interval (8-12 hrs)
- Reflex tachycardia
- Methemoglobinemia (high doses)
Mechanism of GTN: → Releases NO → activates guanylate cyclase → ↑ cGMP → smooth muscle relaxation → venodilation (primarily) + arteriolar dilation at higher doses → ↓ preload (and afterload) → ↓ cardiac work.
SECTION 8: HORMONAL CONTRACEPTIVES
Q: Classification of oral contraceptives?
- Combined oral contraceptives (COC): Estrogen + Progestin
- Monophasic (fixed dose throughout cycle)
- Biphasic, Triphasic (varying doses)
- Progestin-only pill (POP / Minipill): Norethindrone, Levonorgestrel
- Emergency contraception: Levonorgestrel (Plan B), Ulipristal acetate
- Long-acting: DMPA injection (Depo-Provera), Implant (Implanon - etonogestrel)
Q: Mechanism of oral contraceptives?
- Primary (estrogen + progestin): Inhibit GnRH pulsatility → suppress FSH and LH → inhibit ovulation (most important)
- Progestin effect: Thickens cervical mucus → prevents sperm penetration
- Alters endometrium → unfavorable for implantation
- Reduces tubal motility
Q: Adverse effects of COC?
- Nausea, vomiting, breakthrough bleeding (common, usually transient)
- Thromboembolic events (DVT, PE, stroke) - due to estrogen → ↑ clotting factors
- Hypertension
- Weight gain
- Decreased libido
- Breast tenderness
- Chloasma (facial pigmentation)
- Drug interactions (rifampicin, phenytoin reduce efficacy via CYP450 induction)
Q: Contraindications to COC?
- History of DVT, PE, or stroke
- Hypertension
- Smoker >35 years old
- Breast cancer or estrogen-sensitive cancer
- Migraine with aura
- Severe liver disease
- Pregnancy
- Breastfeeding (estrogen reduces milk production)
Q: Why is the minipill (progestin-only) preferred in lactating mothers?
- COC contains estrogen → suppresses prolactin → reduces breast milk production
- Minipill has no estrogen → does not affect lactation
- Progestin-only pill primarily acts by thickening cervical mucus; works without suppressing lactation
- Also excreted in small amounts in breast milk but considered safe
SECTION 9: OBSTETRICS - ANTIHYPERTENSIVES IN PREGNANCY
Q: Drug of choice for pregnancy-induced hypertension (PIH)?
- Mild-moderate PIH: Methyldopa (first-line, safest)
- Alternatives: Labetalol, Nifedipine (long-acting)
- Acute/severe PIH: Labetalol IV or Hydralazine IV
- Magnesium sulfate for eclampsia prevention (not antihypertensive, but prevents seizures)
- Avoid: ACE inhibitors, ARBs (fetotoxic - renal agenesis, oligohydramnios), thiazides
Q: Types of uterine bleeding?
| Type | Description |
|---|
| Menorrhagia | Heavy, regular menstrual bleeding |
| Metrorrhagia | Irregular, between periods |
| Menometrorrhagia | Heavy + irregular |
| Polymenorrhea | Frequent periods (<21 days) |
| Oligomenorrhea | Infrequent periods (>35 days) |
| Dysfunctional uterine bleeding (DUB) | Abnormal bleeding without structural cause |
| Postpartum hemorrhage | Bleeding after delivery |
SECTION 10: OPHTHALMOLOGY
(Covered under Atropine section above)
- Fundus examination: Tropicamide (preferred - short-acting, 4-6 hrs)
- Elderly: Tropicamide or phenylephrine (to avoid angle-closure glaucoma)
- Uveitis: Atropine (long-acting, prevents synechiae)
- Refraction in children: Cyclopentolate (balances duration with cycloplegia needed)
RAPID REVISION: HIGH-YIELD ONE-LINERS
| Topic | Key Point |
|---|
| Succinylcholine + genetics | Atypical pseudocholinesterase → prolonged apnea; dibucaine number |
| ACE inhibitor cough | ↑ bradykinin → use ARB if cough develops |
| Thiazide + uric acid | Compete with urate secretion + volume contraction → hyperuricemia |
| Prazosin 1st dose | Take at bedtime; start 0.5 mg |
| Methyldopa in pregnancy | Drug of choice - central α2 agonist, fetal safe |
| Atropine poisoning | Physostigmine antidote |
| OPC poisoning | Atropine (muscarinic) + Pralidoxime (reactivates AChE) - give early |
| Furosemide vs thiazide Ca | Furo = ↑Ca excretion; Thiazide = ↓Ca excretion |
| Metformin contraindications | GFR <30, acute HF, hypoxic states (lactic acidosis risk) |
| Minipill in lactation | No estrogen → does not reduce milk supply |
| Partial agonist + full agonist | Partial agonist acts as antagonist in presence of full agonist |
| Down-regulation | Chronic agonist → receptor decrease |
| Up-regulation | Chronic antagonist (e.g., beta-blocker) → receptor increase → rebound if stopped suddenly |
| WHO ADR definition | Noxious + unintended + at normal therapeutic doses |
| GLP-1 agonists | Glucose-dependent insulin secretion = no hypoglycemia alone |
| Linagliptin | Only DPP-4 inhibitor safe in severe renal failure without dose adjustment |
| Mydriatic in elderly | Tropicamide (short-acting) - avoids angle-closure glaucoma |
This covers your entire viva list. Let me know which topic you want to drill deeper, practice in Q&A format, or get examiner-style grilling on - and we'll go topic by topic from here.