Explain me aldosterone escape
Reading File
Finding Sources
Searching PubMed
"aldosterone escape" mechanism
Reading File
Reading File
Reading Article
Reading File
Loading Image
Loading Image
I now have comprehensive material to write a thorough explanation. Here is the full answer:
Aldosterone Escape
Definition
Aldosterone escape is the spontaneous return of renal sodium (and water) excretion back to normal after a period of initial sodium retention caused by excess aldosterone - even though aldosterone levels remain elevated. The body "escapes" from the sodium-retaining effects of aldosterone without a fall in aldosterone itself.
It applies to primary aldosteronism (Conn's syndrome). It does NOT occur in states of secondary aldosteronism (heart failure, cirrhosis, nephrotic syndrome) - an important distinction explained below.
The Sequence of Events
1. Initial sodium retention
When aldosterone is in excess, it stimulates principal cells of the collecting duct (and distal tubule) to upregulate epithelial sodium channels (ENaC) and Na+/K+-ATPase. Sodium is reabsorbed, potassium is excreted. Water follows osmotically.
2. Volume expansion
Extracellular fluid (ECF) volume expands by approximately 5-15% above normal. Despite marked sodium retention, plasma sodium concentration rises only slightly because water is retained proportionally (via osmotic drive and ADH).
3. Rise in arterial pressure
The ECF expansion increases venous return, cardiac output, and mean arterial pressure - typically by 15-25 mmHg. This is the key trigger for escape.
4. Pressure natriuresis and pressure diuresis
The elevated blood pressure directly forces more sodium and water out through the kidney - a mechanism called pressure natriuresis/diuresis. This is the single most important driver of escape.
5. Counter-regulatory hormones
Simultaneously, two other mechanisms reinforce the natriuresis:
- Suppression of angiotensin II: Volume expansion suppresses renin, which reduces Ang II. This decreases proximal tubular sodium reabsorption and lowers aldosterone stimulation (though aldosterone remains high in primary aldosteronism due to autonomous secretion).
- Atrial natriuretic peptide (ANP) release: Atrial stretch from volume overload triggers ANP release, which inhibits sodium reabsorption in the collecting duct and promotes natriuresis.
6. New steady state
After ~1-2 days, urinary sodium excretion returns to equal sodium intake. Sodium balance is re-established. The ECF remains modestly expanded and hypertension persists (as long as aldosterone remains elevated), but no further sodium accumulation occurs.
- Guyton and Hall Textbook of Medical Physiology
This is beautifully illustrated in the experimental data below, where aldosterone was infused at ~20x normal levels in dogs:
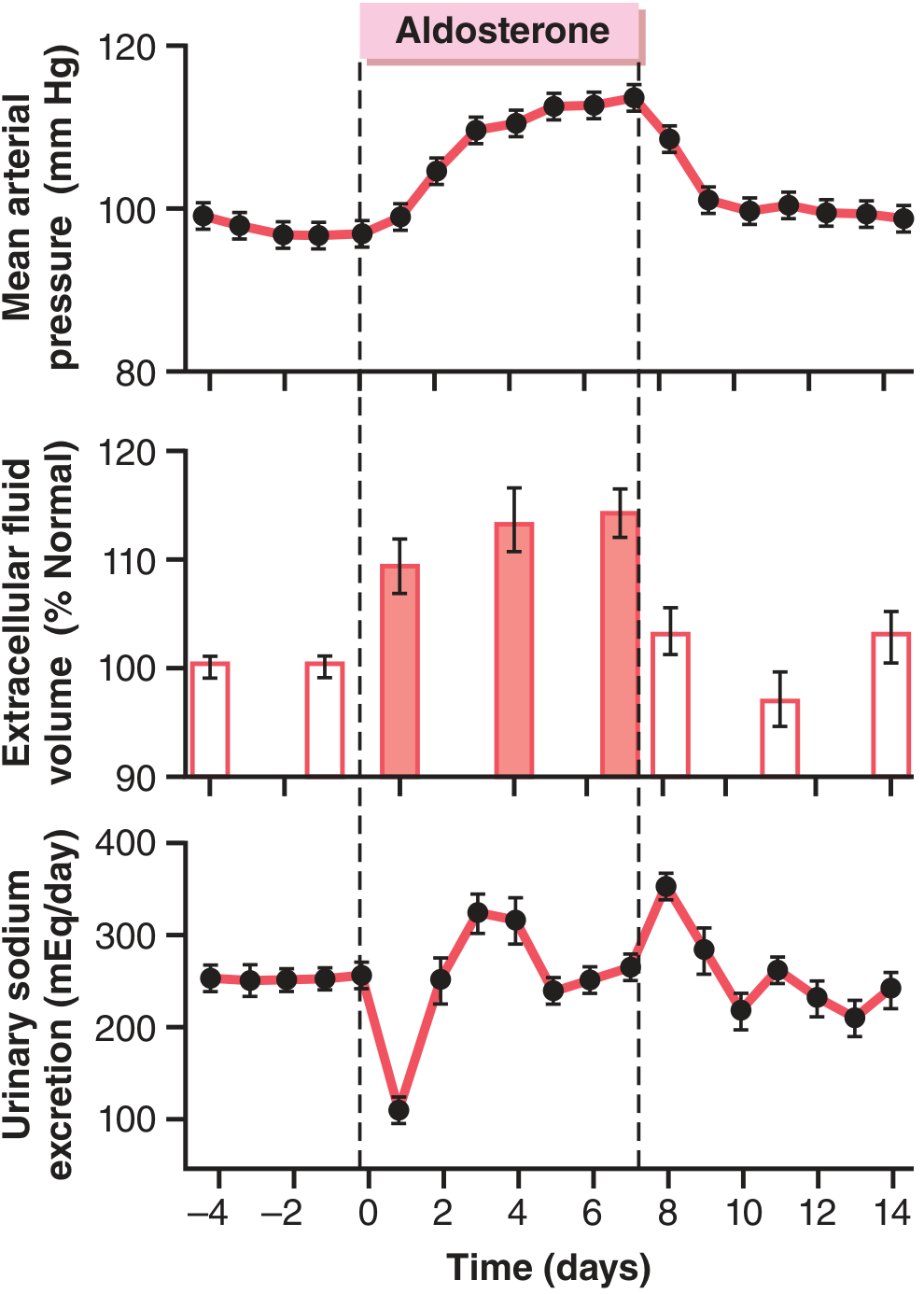
Figure 78.3 from Guyton & Hall: Note urinary sodium drops sharply on day 1, then returns to baseline by day 2-3 as arterial pressure rises - this is aldosterone escape.
Mechanisms Summary Table
| Mechanism | How it causes natriuresis |
|---|---|
| Pressure natriuresis (primary) | Elevated BP directly increases urinary Na+ excretion |
| ANP release | Atrial stretch → ANP → inhibits collecting duct Na+ reabsorption |
| Angiotensin II suppression | Volume expansion suppresses renin → less proximal Na+ reabsorption |
| WNK kinase system (emerging) | Hypokalemia from aldosterone activates WNK1/4 → modulates NCC in distal tubule (recently described in the 2026 JASN review) |
The NCC (thiazide-sensitive Na-Cl cotransporter) has also been identified as a mediator - its downregulation during escape contributes to distal sodium delivery. - Comprehensive Clinical Nephrology, 7th Ed.
Why Escape Does NOT Occur in Secondary Aldosteronism
This is a high-yield distinction. In conditions like heart failure, cirrhosis, and nephrotic syndrome, aldosterone is elevated secondarily due to effective arterial underfilling. These patients do NOT escape and instead accumulate edema. The reason:
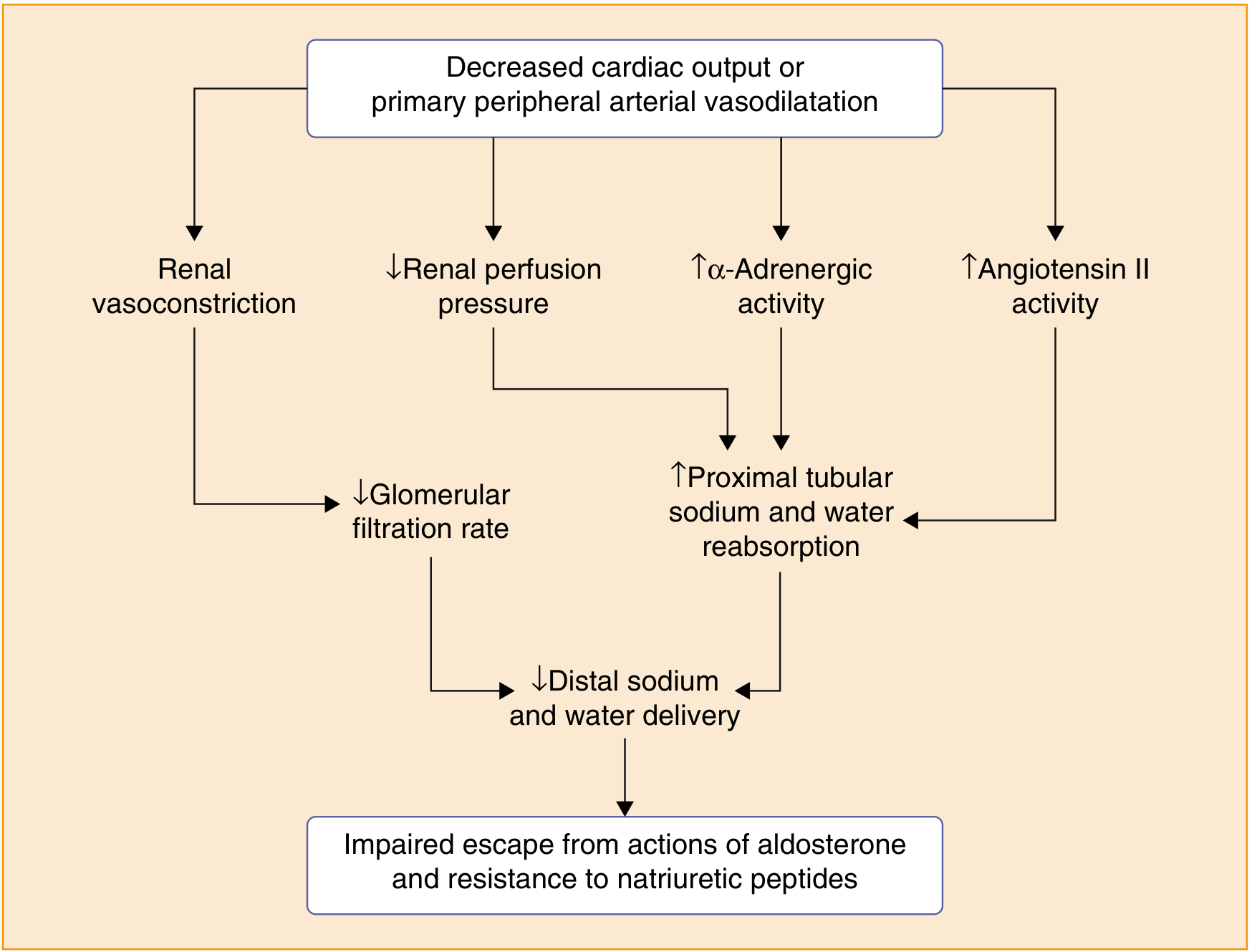
From Goldman-Cecil Medicine: In heart failure/cirrhosis, decreased cardiac output or peripheral vasodilation causes renal vasoconstriction, reduced GFR, and increased proximal tubular Na+ reabsorption - so less Na+ reaches the distal tubule where aldosterone acts, making escape impossible.
In these states:
- Arterial pressure does not rise enough to trigger pressure natriuresis (or cannot because of impaired cardiac output)
- ANP effects are blunted
- Reduced distal sodium delivery means there is little sodium left to escape with
- The RAAS remains chronically activated
"Aldosterone Escape" in the Context of ACE Inhibitors / ARBs
There is a second (confusingly named) use of the term in pharmacology: when patients on ACE inhibitors or ARBs initially have reduced aldosterone, but after weeks to months, aldosterone levels rise back toward baseline. This is sometimes called the "aldosterone breakthrough" or pharmacological aldosterone escape. It occurs because:
- Non-ACE pathways (e.g., chymase) continue to generate Ang II
- Renin levels compensatorily rise
This is clinically relevant in heart failure and CKD, where incomplete aldosterone blockade despite RAAS therapy can lead to ongoing organ damage. - Goodman & Gilman's Pharmacological Basis of Therapeutics
Key Points to Remember
| Feature | Value |
|---|---|
| Occurs in | Primary aldosteronism |
| Does NOT occur in | Secondary aldosteronism (HF, cirrhosis, nephrotic syndrome) |
| Primary mechanism | Pressure natriuresis (arterial BP rise forces Na+ excretion) |
| Supporting mechanisms | ANP release, Ang II suppression, WNK/NCC modulation |
| End result | Na+ balance restored, but hypertension and hypokalemia persist |
| Time course | ~1-2 days after aldosterone elevation |
| No escape explains | Edema accumulation in HF/cirrhosis despite RAAS activation |
Recent evidence note: A 2026 review in JASN (PMID 41056081) re-examines aldosterone escape mechanisms, highlighting the WNK kinase system as a potassium-sensitive distal homeostasis pathway that may contribute to the escape phenomenon - suggesting the full mechanism is still being worked out.
This is a shared conversation. Sign in to Orris to start your own chat.