Myocardial Infarction
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND "management"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have comprehensive data from multiple authoritative textbooks. Let me compile the full answer.
Myocardial Infarction (MI)
Definition
Myocardial infarction (MI), commonly called a "heart attack," is necrosis of heart muscle resulting from ischemia. The 2018 joint task force of U.S. and European Cardiology groups defines MI as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia." - Robbins & Kumar Basic Pathology, p. 354
Epidemiology & Risk Factors
- The frequency of MI rises progressively with age and with increasing risk factors for atherosclerosis
- ~10% of MIs occur before age 40; ~45% before age 65
- Men are at greater risk than women, though this gap narrows with age
- Women are relatively protected during reproductive years; menopause (with declining estrogen) is associated with accelerated coronary artery disease
- IHD is the most common cause of death in older women
Pathogenesis
Coronary Artery Occlusion - The Sequence of Events
The vast majority of MIs are caused by acute thrombosis within coronary arteries. The typical sequence:
- Plaque disruption - An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces, exposing subendothelial collagen and necrotic plaque contents to blood
- Platelet activation - Platelets adhere, aggregate, and release thromboxane A₂, ADP, and serotonin, causing further aggregation and vasospasm
- Coagulation cascade - Exposure of tissue factor adds to the growing thrombus
- Complete occlusion - Within minutes, the thrombus may fully occlude the coronary artery lumen
In ~10% of MIs, transmural infarction occurs in the absence of occlusive atherosclerosis - such cases are attributed to coronary vasospasm, embolization from mural thrombi (e.g., in atrial fibrillation), or valve vegetations.
Angiography performed within 4 hours of MI onset demonstrates coronary thrombosis in ~90% of cases. By 12-24 hours, evidence of thrombosis is seen in only 60%, even without intervention - suggesting some occlusions clear spontaneously.
- Robbins & Kumar Basic Pathology, p. 354
Myocardial Response to Ischemia
Within seconds: Aerobic metabolism ceases → ATP drops → lactic acid accumulates
Within minutes: Rapid loss of contractility
20-40 minutes: Irreversible damage and coagulative necrosis begins
Earliest sign of necrosis: Disruption of sarcolemmal membrane integrity → intracellular macromolecules (troponins, CK-MB) leak into circulation
Within minutes: Rapid loss of contractility
20-40 minutes: Irreversible damage and coagulative necrosis begins
Earliest sign of necrosis: Disruption of sarcolemmal membrane integrity → intracellular macromolecules (troponins, CK-MB) leak into circulation
The subendocardial zone is the first and most vulnerable region because:
- It is the last area to receive blood from epicardial vessels
- It is exposed to relatively high intramural pressures impeding inflow
- Oxygen requirement is highest here (~1.3 mL O₂/100g/min just to remain viable)
With more prolonged ischemia, a wavefront of cell death moves outward from the subendocardium toward the epicardium, driven by progressive tissue edema and reactive oxygen species.
- Robbins & Kumar Basic Pathology, p. 355; Guyton & Hall Medical Physiology, p. 271
Classification: STEMI vs. NSTEMI
| Feature | STEMI | NSTEMI |
|---|---|---|
| Coronary occlusion | Complete | Partial / incomplete |
| Infarct depth | Transmural | Subendocardial |
| ECG | ST elevation + Q waves | ST depression or T-wave inversion, NO Q waves |
| Mortality (in-hospital) | ~10% | ~6% |
| Management | Urgent reperfusion (PCI/thrombolysis) | Conservative or early invasive |
- Robbins & Kumar Basic Pathology, p. 359
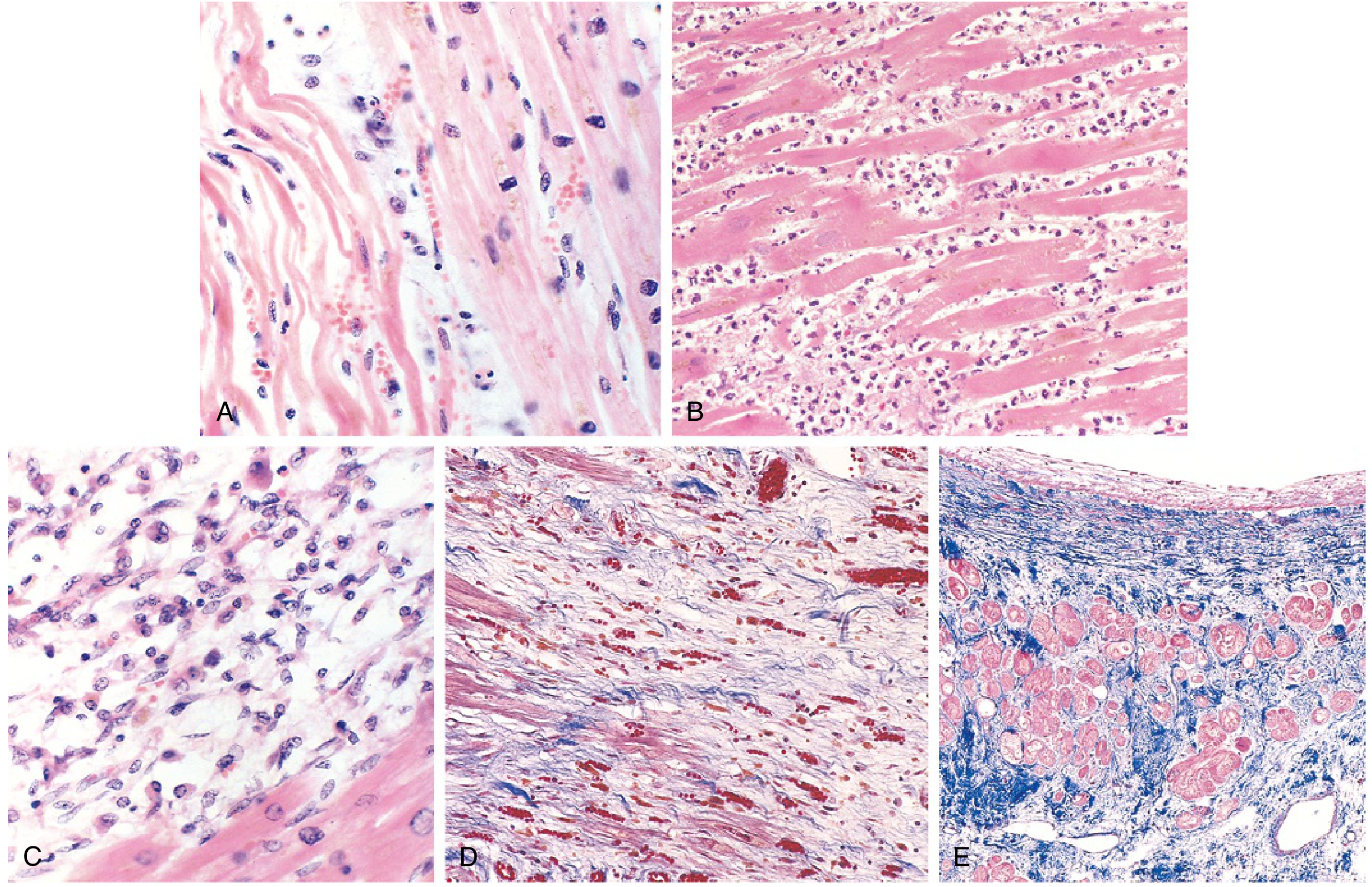
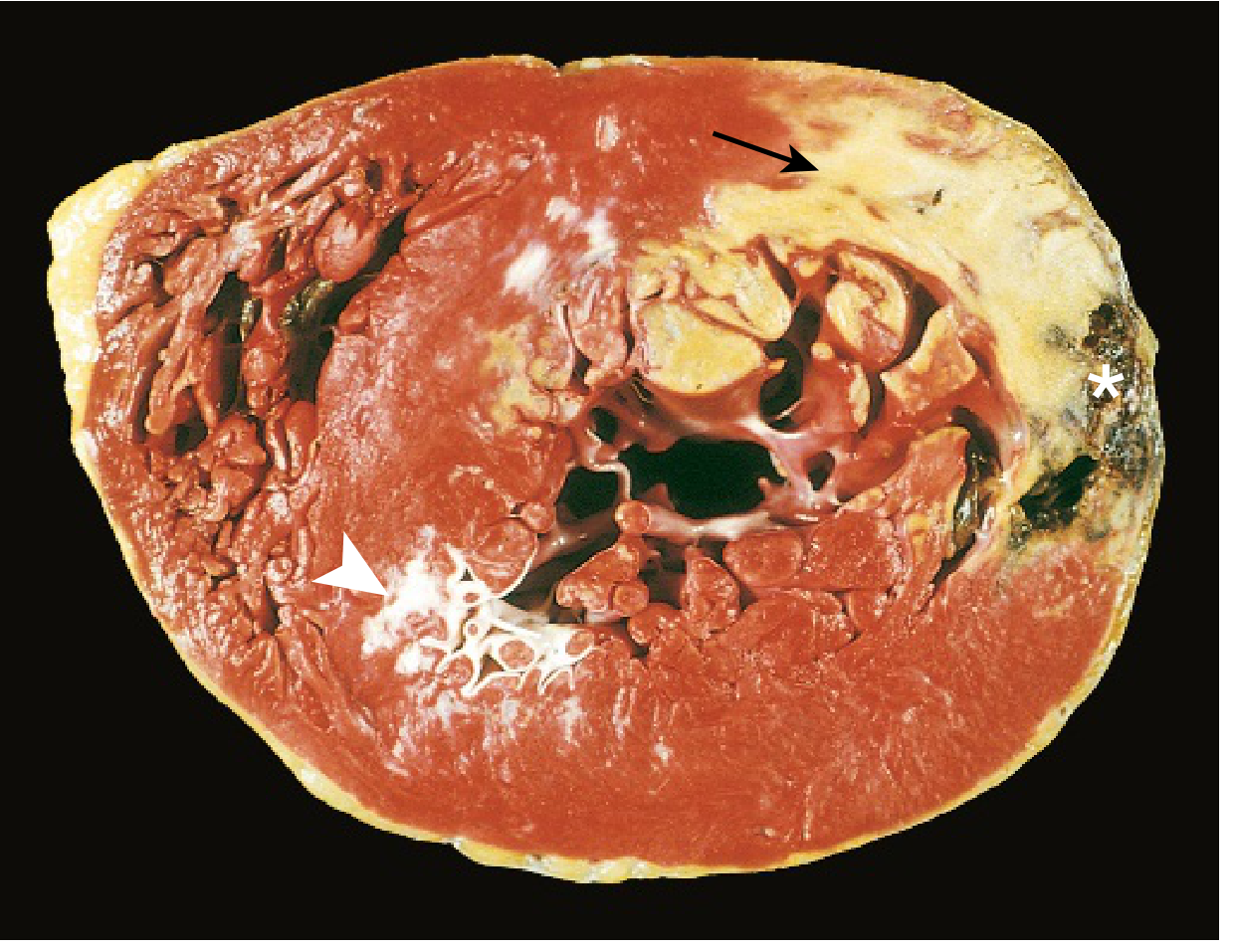
Morphology - Time-Dependent Changes
The gross and microscopic appearance of MI follows a highly characteristic sequence:
| Time | Gross Features | Light Microscopy |
|---|---|---|
| 0-30 min | None | None (reversible) |
| 30 min - 4 hr | None | Variable waviness of fibers at border |
| 4-12 hr | Occasional dark mottling | Onset of coagulative necrosis; edema; hemorrhage |
| 12-24 hr | Dark mottling | Coagulative necrosis; pyknotic nuclei; hypereosinophilic myocytes; contraction band necrosis; early neutrophil infiltrate |
| 1-3 days | Mottling with yellow-tan infarct center | Coagulative necrosis with loss of nuclei and striations; increased neutrophil infiltrate |
| 7-10 days | Maximally yellow-tan, soft | Nearly complete removal of necrotic myocytes by macrophages; granulation tissue at margins |
| Weeks | Gray-white scar forms | Fibrosis / scar replacing necrotic tissue |
Key diagnostic point: Infarcts <12 hours old are usually NOT grossly apparent. Vital stains like triphenyl tetrazolium chloride (TTC) can reveal infarcts >3 hours old - the infarcted area remains unstained (pale) because the enzyme LDH has leaked out of dead cells. Old scars appear white and glistening.
ECG Changes
Three major electrical abnormalities occur in acute MI (from Ganong's Medical Physiology):
| Defect in Infarcted Cells | Current Flow | ECG Change |
|---|---|---|
| Rapid repolarization (accelerated K⁺ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential (loss of intracellular K⁺) | Into infarct | TQ depression → appears as ST elevation |
| Delayed depolarization | Out of infarct | ST segment elevation |
Evolution of ECG changes:
- Acute: ST elevation in leads overlying the infarct; reciprocal ST depression in opposite leads
- Days to weeks: ST changes subside; dead muscle becomes electrically silent → Q waves appear (pathological Q = >1 mm wide, >1/4 of QRS height)
- "Non-Q-wave" infarcts (NSTEMI) tend to be less severe but carry a higher risk of reinfarction
Localization by leads:
- Anterior MI: V1-V4 (LAD territory)
- Inferior MI: II, III, aVF (RCA territory)
- Lateral MI: I, aVL, V5-V6 (LCx territory)
- Posterior MI: tall R in V1-V2 (mirror image)
- Ganong's Review of Medical Physiology, p. 534
Cardiac Biomarkers
The laboratory diagnosis of MI relies on measuring intracellular proteins that leak through damaged sarcolemmal membranes:
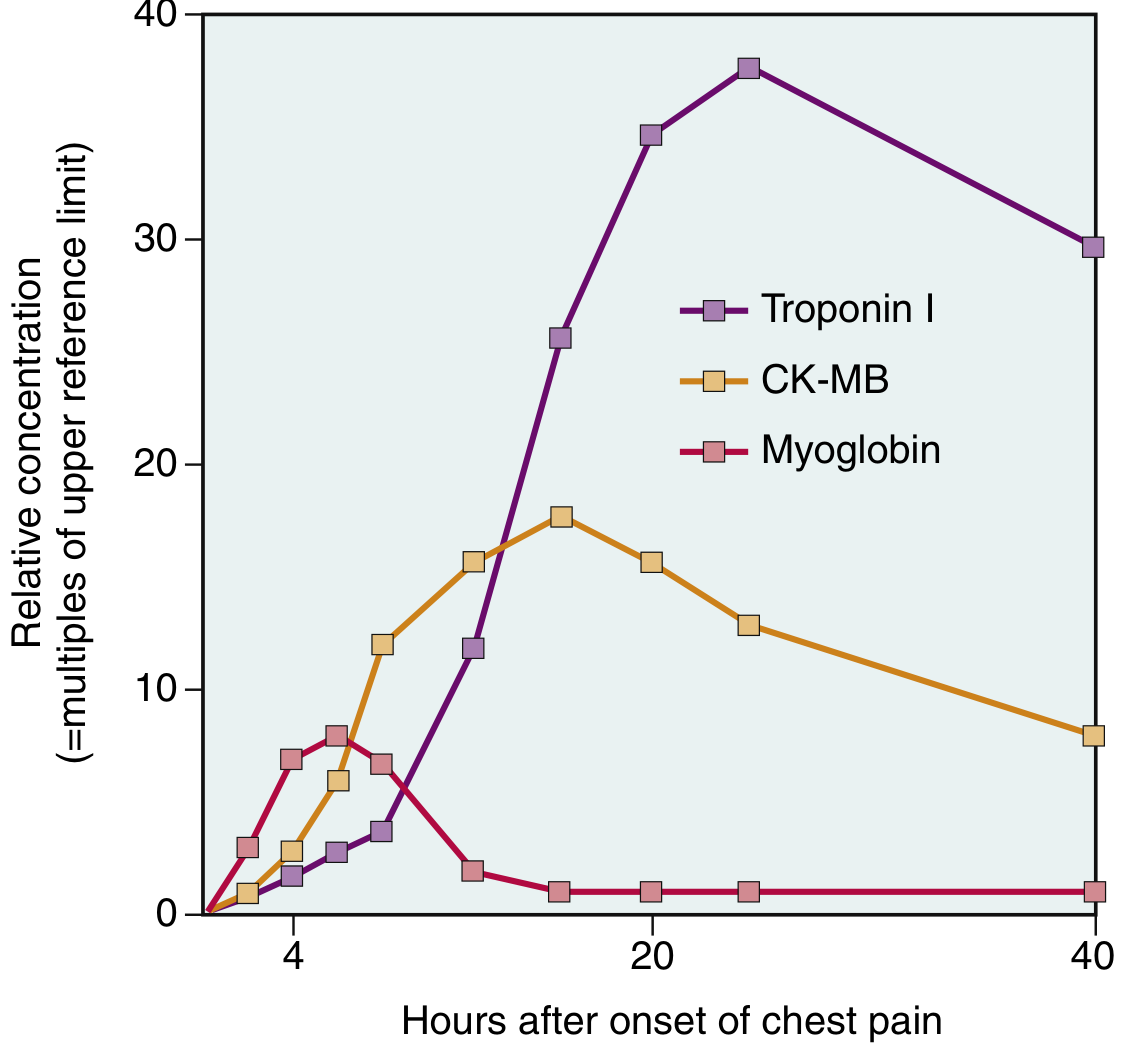
| Biomarker | Rise | Peak | Return to Normal | Notes |
|---|---|---|---|---|
| Troponin I / T | 2-4 hr | 48 hr | 7-10 days | Gold standard; highest sensitivity and specificity |
| CK-MB | 2-4 hr | 24-48 hr | ~72 hr | Useful for detecting reinfarction |
| Myoglobin | 1-2 hr | 4-8 hr | 24 hr | Earliest but non-specific |
TnI and TnT are normally undetectable in circulation. After reperfusion, both troponin and CK-MB levels peak earlier due to rapid washout from necrotic tissue.
- Robbins & Kumar Basic Pathology, p. 359
Clinical Features
Classic presentation:
- Severe, crushing substernal chest pain (pressure/tightness) radiating to neck, jaw, epigastrium, or left arm
- Pain lasts minutes to hours - NOT relieved by nitroglycerin or rest
- Diaphoresis, nausea, dyspnea, rapid and weak pulse
Atypical / "Silent" MI (~25% of cases):
- Particularly common in diabetics (autonomic neuropathy blunts pain perception) and elderly
- May present as jaw pain, epigastric discomfort, or sudden dyspnea only
With massive MI (>40% of LV):
- Cardiogenic shock develops (BP < 90 mmHg, poor perfusion, pulmonary edema)
- Robbins & Kumar Basic Pathology, p. 358
Complications
Nearly three-fourths of patients with acute MI experience one or more complications:
Potentially Lethal Complications
| Complication | Timing | Notes |
|---|---|---|
| Ventricular free wall rupture | 3-7 days post-MI | Fatal hemopericardium + cardiac tamponade; least common but most deadly |
| Interventricular septal rupture | 3-7 days | Creates acute VSD; new harsh holosystolic murmur |
| Papillary muscle rupture | 3-7 days | Acute severe mitral regurgitation |
Rupture occurs 3-7 days post-MI - the time when lysis of necrotic myocardium is maximal and granulation tissue is soft and friable.
Other Complications
- Contractile dysfunction / Cardiogenic shock - LV failure proportional to infarct size; ~10% of transmural MIs
- Arrhythmias - ~90% of patients develop some rhythm disturbance; ventricular fibrillation risk highest in the first hour (accounts for most pre-hospital deaths); includes heart block, VT, VF, SVT
- Pericarditis - Fibrinohemorrhagic pericarditis days 2-3, resolves in 1-2 weeks; presents with anterior chest pain and pericardial friction rub; Dressler syndrome = late autoimmune pericarditis (weeks to months later)
- Infarct extension - Additional necrosis at infarct margins in the days after MI
- Mural thrombus - Overlying the infarcted endocardium, risk of systemic embolism
- Ventricular aneurysm - LV dilation and wall thinning, especially anterior apical infarcts; paradoxical wall motion on echo; risk of thrombus, arrhythmia, HF
- Progressive heart failure - Due to loss of functional myocardium
- Robbins & Kumar Basic Pathology, p. 360; Guyton & Hall Medical Physiology
Causes of Death After Acute MI
Per Guyton & Hall Medical Physiology, the four major causes:
- Decreased cardiac output - Systolic stretch (paradoxical bulging of infarcted area) wastes ventricular energy → cardiogenic shock
- Pulmonary edema - Damming of blood in pulmonary circulation from LV failure
- Ventricular fibrillation - Accounts for 80-90% of cardiac deaths in ischemia setting; highest risk in first hour
- Cardiac rupture - Rare, usually fatal
Management Principles
Immediate goals (time-critical):
- STEMI: Urgent reperfusion within 90 minutes of first medical contact - primary PCI (percutaneous coronary intervention) is preferred; thrombolysis if PCI not available
- NSTEMI/UA: Anticoagulation, antiplatelet therapy, risk stratification for early invasive vs. conservative strategy
Acute pharmacotherapy (MONA + antiplatelet):
- Aspirin (antiplatelet) - immediately
- P2Y12 inhibitor (clopidogrel, ticagrelor, prasugrel) - dual antiplatelet therapy
- Anticoagulation (UFH, LMWH, fondaparinux, bivalirudin)
- Beta-blockers - reduce myocardial oxygen demand, antiarrhythmic
- ACE inhibitors / ARBs - reduce ventricular remodeling, especially with reduced EF
- Statins - plaque stabilization, reduce recurrence
- Oxygen - only if SpO₂ < 90%
- Morphine - for refractory pain (use with caution - some evidence of harm in NSTEMI)
- Nitrates - for pain relief, but avoid in RV infarction, hypotension, or recent PDE5 inhibitor use
Long-term:
- Dual antiplatelet therapy (1 year post-stent)
- Beta-blocker, ACE inhibitor/ARB, statin - indefinite
- Cardiac rehabilitation
- Risk factor modification
Prognosis
- Overall in-hospital mortality: ~7-8%
- STEMI in-hospital mortality: ~10%
- NSTEMI in-hospital mortality: ~6%
- Out-of-hospital STEMI mortality: One-third die, usually from arrhythmia within the first hour before receiving medical care
- With better and earlier in-hospital care, the STEMI-NSTEMI mortality gap is narrowing
Sources:
- Robbins & Kumar Basic Pathology, 10th ed., pp. 354-360
- Guyton and Hall Textbook of Medical Physiology, pp. 271-272
- Ganong's Review of Medical Physiology, 26th ed., pp. 534-536
- Braunwald's Heart Disease, 12th ed.
This is a shared conversation. Sign in to Orris to start your own chat.