Classify intraepidermal immunobullous diseases.Describe management of Pemphigus vulgaris in 10 marks format for 3rd yr dermatology post graduate theory examination
Reading File
Finding Sources
Finding Sources
Searching PubMed
pemphigus vulgaris management treatment
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Searching Images
pemphigus vulgaris intraepidermal blister acantholysis histology
Loading Image
I now have all the information needed for a comprehensive, exam-style answer. Here is the complete response:
Classification of Intraepidermal Immunobullous Diseases
Intraepidermal immunobullous diseases (also called intraepidermal blistering diseases with autoantibodies) are grouped as follows:
A. Pemphigus Group
| Disease | Level of Cleavage | Target Antigen | Key Feature |
|---|---|---|---|
| Pemphigus Vulgaris (PV) | Suprabasal | Desmoglein 3 (± Dsg1) | Most common; oral mucosa first |
| Pemphigus Vegetans | Suprabasal | Desmoglein 3 | Variant of PV; verrucous plaques in flexures |
| Pemphigus Foliaceus (PF) | Subcorneal/granular | Desmoglein 1 | Superficial; no mucosal involvement |
| Pemphigus Erythematosus | Subcorneal | Desmoglein 1 + ANA | Variant of PF; sebaceous distribution |
| Endemic Pemphigus Foliaceus (Fogo Selvagem) | Subcorneal | Desmoglein 1 | Endemic in Brazil; triggered by insect bite |
| Paraneoplastic Pemphigus (PNP) | Suprabasal ± lichenoid | Desmoplakins, periplakin, envoplakin, BP230 | Associated with lymphoma, thymoma; severe mucosal involvement |
| Drug-induced Pemphigus | Variable | Dsg1 or Dsg3 | Triggered by thiols (penicillamine, captopril) |
B. IgA Pemphigus
A distinct subset with IgA (not IgG) autoantibodies; two subtypes:
| Subtype | Target | Histology |
|---|---|---|
| Subcorneal Pustular Dermatosis (SPD) type | Desmocollin 1 | Subcorneal neutrophilic pustules |
| Intraepidermal Neutrophilic (IEN) type | Desmoglein 1 or 3 | Mid-epidermal neutrophilic pustules |
Management of Pemphigus Vulgaris — 10 Marks
Introduction
Pemphigus Vulgaris (PV) is a potentially life-threatening autoimmune blistering disease caused by IgG autoantibodies against desmoglein 3 (mucous membrane type) and desmoglein 1 + 3 (mucocutaneous type), leading to suprabasal acantholysis. Management aims to suppress autoantibody production, control disease, taper steroids, and prevent complications.
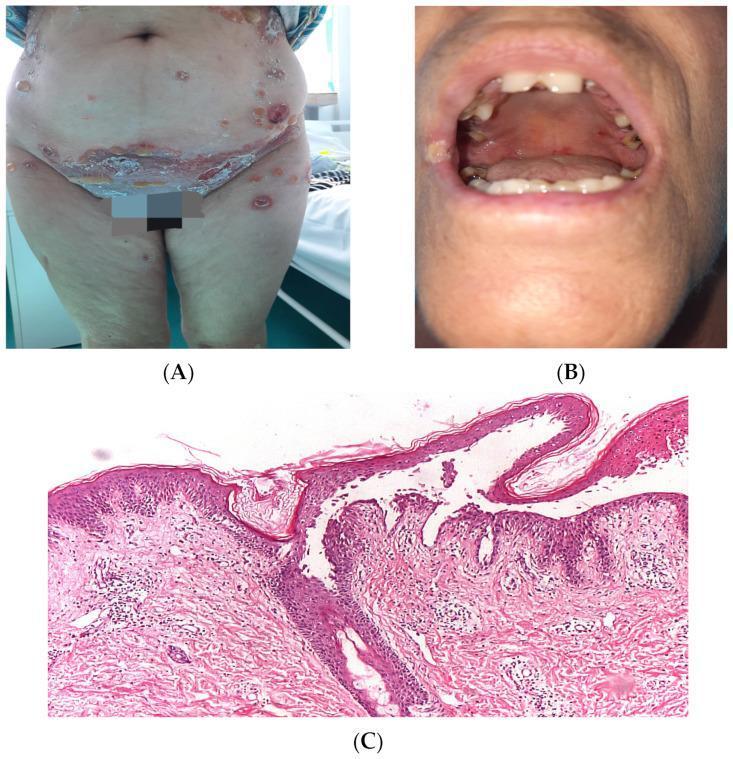
1. Assessment Before Treatment
- Disease severity scoring: Pemphigus Disease Area Index (PDAI) or Autoimmune Bullous Skin disorder Intensity Score (ABSIS)
- Baseline investigations: CBC, LFT, RFT, blood glucose, urine for cyclophosphamide users, hepatitis B/C serology, quantitative anti-Dsg3/Dsg1 ELISA titers, chest X-ray, bone densitometry
- TPMT enzyme assay before azathioprine initiation
2. General/Supportive Measures
- Wound care: non-adherent dressings, topical antiseptics (dilute potassium permanganate soaks), Vaseline gauze to raw areas
- Oral care: antiseptic mouthwashes (chlorhexidine), viscous lignocaine for pain relief
- Nutritional support: high-protein diet; nasogastric feeding if severe oral involvement precludes eating
- Prevention of secondary infection: systemic antibiotics when superinfection occurs
- Avoid triggering drugs (ACE inhibitors, NSAIDs, thiol-containing drugs) and trauma
3. First-Line: Systemic Corticosteroids
Systemic corticosteroids are the mainstay of therapy.
- Oral prednisolone: 1 mg/kg/day (usually 60–100 mg/day) as starting dose
- Continue until no new lesions for ≥2 weeks and >80% of existing lesions healed
- Then taper slowly (by 25% every 4–6 weeks) to a maintenance dose (~5–10 mg/day)
- Pulse methylprednisolone (1 g/day IV for 3–5 consecutive days) is reserved for severe, rapidly progressive disease
4. Adjuvant Steroid-Sparing Immunosuppressants
Used in combination with corticosteroids to achieve disease control and reduce steroid dose/side effects:
| Agent | Dose | Notes |
|---|---|---|
| Azathioprine | 2–4 mg/kg/day (100–300 mg/day) | First-line adjuvant; check TPMT; risk: myelosuppression, hepatotoxicity |
| Mycophenolate Mofetil (MMF) | 2–3 g/day | Similar efficacy to azathioprine; more GI side effects; preferred in liver disease |
| Cyclophosphamide | 1–3 mg/kg/day oral OR 500–1000 mg/m² IV pulse every 4 weeks | For refractory cases; risk: hemorrhagic cystitis, infertility, bladder carcinoma |
| Methotrexate | 7.5–20 mg/week | Used in milder disease; hepatotoxicity |
| Dapsone | 50–150 mg/day | Adjuvant in IgA pemphigus |
Tapering strategy: Once complete remission is achieved with combination therapy, taper prednisolone first; when dose reaches 5–10 mg/day, gradually discontinue the immunosuppressant.
5. Rituximab (B-Cell Depletion Therapy)
Rituximab — a chimeric anti-CD20 monoclonal antibody — is now considered a first-line treatment for moderate-to-severe PV alongside short-term steroids (based on the 2017 Ritux 3 RCT, NEJM).
Mechanism: Depletes CD20+ B cells → reduces anti-Dsg IgG autoantibodies and desmoglein-specific T cells
Dosing regimens:
- Lymphoma protocol: 375 mg/m² IV once weekly × 4 doses; may repeat every 3–6 months
- Rheumatoid arthritis protocol: 1 g IV initially, then 1 g at 2 weeks (preferred in recent practice)
Role: First-line in moderate-severe PV; for steroid-refractory cases; allows significant steroid dose reduction
Side effects: Infusion reactions, increased infection risk (especially PML with JC virus), hypogammaglobulinemia; screen for hepatitis B reactivation before use
⚠️ Recent evidence update (2025): A systematic review and meta-analysis (PMID 40787442) confirmed that low-dose rituximab is efficacious and safe in pemphigus, supporting its use in resource-limited settings.
6. Third-Line / Rescue Therapies
High-Dose Intravenous Immunoglobulin (IVIg)
- Dose: 400 mg/kg/day for 5 consecutive days; may be repeated monthly
- Mechanism: Accelerates clearance of pathogenic IgG via Fc receptor saturation; provides anti-idiotypic antibodies; immunomodulatory
- Indication: Steroid-resistant/refractory PV; useful when immunosuppression is contraindicated (pregnancy, active infection)
- Evidence: Multicenter randomized placebo-controlled trial demonstrated safety and efficacy
Plasmapheresis / Immunoadsorption
- Rapidly reduces circulating autoantibody titers
- Useful in severe acute PV with high antibody titers
- Must be combined with immunosuppression (prednisolone + cyclophosphamide) to prevent post-pheresis autoantibody rebound
- 1–2 sessions per week initially
Extracorporeal Photopheresis
- 2 days per month
- Reserved for refractory cases
7. Topical Therapy (Adjunctive)
- Topical Class I corticosteroids (clobetasol propionate): for localized persistent lesions
- Topical antibiotics: prevent secondary infection
- Topical immunomodulators: cyclosporine mouthwash for oral lesions, tacrolimus ointment for localized disease
- Intralesional corticosteroids: for persistent isolated lesions
- Intralesional rituximab (5 mg/cm²): investigational for refractory localized lesions
8. Monitoring and Disease Activity Assessment
- Clinical: number of new blisters/erosions, mucosal activity (PDAI score)
- Serological: quantitative anti-Dsg3 and anti-Dsg1 ELISA — titers correlate with disease activity
- Monitor for side effects: blood glucose, blood pressure, weight, bone density (DEXA scan), ophthalmology review (cataract, glaucoma screening with long-term steroids)
- Prophylaxis during immunosuppression:
- PCP prophylaxis: co-trimoxazole
- Pneumococcal, influenza vaccines (before starting immunosuppression)
- Calcium + Vitamin D + bisphosphonate for corticosteroid-induced osteoporosis prevention
- PPI for GI protection
9. Defining Remission and Endpoint
- Complete remission on therapy: No new lesions for ≥2 months while on minimal therapy (≤10 mg/day prednisone)
- Complete remission off therapy: No new lesions for ≥2 months after discontinuation of all therapy
- Continue monitoring anti-Dsg titers even after clinical remission — rising titers predict relapse
10. Management Summary (Therapeutic Ladder)
MILD-MODERATE PV
→ Prednisolone 0.5–1 mg/kg/day ± Azathioprine or MMF
→ Topical steroids as adjuvant
MODERATE-SEVERE PV (or rapid progression)
→ Rituximab (1g × 2 doses at 0 and 2 weeks) + short-term prednisolone (0.5 mg/kg/day)
→ OR: Prednisolone 1 mg/kg/day + Azathioprine/MMF/Cyclophosphamide
REFRACTORY PV
→ High-dose IVIg (400 mg/kg/day × 5 days monthly)
→ Plasmapheresis + immunosuppression
→ Extracorporeal photopheresis
→ Clinical trial consideration (e.g., BTK inhibitors, neonatal Fc receptor blockers — efgartigimod)
Prognosis
Before systemic steroids: mortality was ~75%. Currently, mortality has fallen to <5%, primarily from treatment complications (infections, steroid side effects) rather than the disease itself. Rituximab as first-line therapy has further improved outcomes with sustained remissions and reduced steroid exposure.
Sources: Dermatology 2-Volume Set 5e (Bolgnia); Fitzpatrick's Dermatology Vol 1–2; Robbins & Kumar Basic Pathology — Dermatology 2-Volume Set 5e, pp. 619–622
Write short notes on -a)cutaneous lymphoid hyperplasia and b)Rosacea
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Searching Images
rosacea subtypes erythematotelangiectatic papulopustular rhinophyma clinical
Reading File
Searching Images
cutaneous lymphoid hyperplasia pseudolymphoma histology germinal center skin
I now have excellent content for both topics. Let me compile the comprehensive short notes.
Short Notes
a) Cutaneous Lymphoid Hyperplasia (CLH)
Definition & Synonyms
Cutaneous lymphoid hyperplasia (CLH), also called pseudolymphoma of the skin, is a benign, reactive, polyclonal lymphoid proliferation in the skin that clinically and/or histologically mimics cutaneous lymphoma. Synonyms include:
- Lymphocytoma cutis
- Lymphadenosis benigna cutis (LABC)
- Spiegler–Fendt sarcoid
- Miliary lymphocytoma
Etiopathogenesis
CLH does not represent a single disease but an exaggerated local immunologic reaction to various stimuli — often unrecognized. A hapten-driven immunologic response to cells damaged by a direct toxic stimulus is postulated.
Inciting agents include:
| Category | Examples |
|---|---|
| Arthropod bites | Ticks (Borrelia in Europe), mites, mosquitoes |
| Infections | Herpes zoster, Lyme borreliosis (B. burgdorferi) |
| Physical agents | Tattoo pigments, metal implants, acupuncture needles, zippers |
| Contact allergens | Nickel, gold |
| Vaccinations | Hepatitis B, influenza, HPV, varicella-zoster |
| Medications | Anticonvulsants (phenytoin, carbamazepine), antihypertensives (ACE inhibitors, β-blockers), antihistamines (cimetidine), biologics (adalimumab, infliximab), NSAIDs, antidepressants, antipsychotics |
Clinical Features
- Morphology: Single (occasionally multiple), firm, erythematous to violaceous papule, nodule, or plaque, 1–3 cm in diameter
- Site: Head, neck, face (especially eyelids, cheeks, earlobes), upper extremities
- Surface: Usually smooth, without scale
- Variants: Multiple clustered papules; panniculitis-like deeper nodules
- Borrelia-associated: Often solitary on the ear, breast, or scrotum/nipple (lymphocytoma cutis)
- Drug-induced: May be widespread; may resemble mycosis fungoides
Histopathology
Two patterns:
B Cell-Predominant (most common)
- Superficial and deep nodular or diffuse dermal infiltrate of lymphocytes, histiocytes, plasma cells, and eosinophils
- Reactive germinal centers with tangible-body macrophages ("tingible body macrophages" = sign of high turnover — reactive rather than neoplastic)
- Polyclonal B cells (no Ig gene rearrangement)
- IHC: Mixed CD20+ and CD3+ populations; bcl-2 negative in germinal center cells (unlike follicular lymphoma)
T Cell-Predominant (usually drug-induced)
- CD4+ T helper lymphocytes in dermis ± CD8+ cytotoxic T cells
- May show epidermotropism resembling mycosis fungoides
- Usually polyclonal TCR gene rearrangement
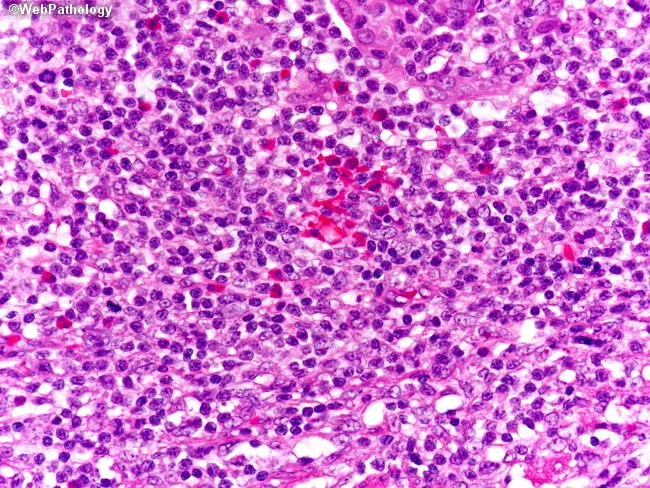
Differential Diagnosis
| Feature | CLH (Pseudolymphoma) | Cutaneous B-Cell Lymphoma |
|---|---|---|
| Lesions | Usually solitary | Often multiple |
| Infiltrate | Mixed (B + T cells) | Monomorphous |
| Germinal centers | Present, reactive | Absent or atypical |
| Clonality | Polyclonal | Monoclonal (IgH rearrangement) |
| bcl-2 in germinal centers | Negative | Positive (follicle center lymphoma) |
| Clinical course | Resolves spontaneously or with treatment | Persistent/progressive |
Also distinguish from: mycosis fungoides (T-cell, epidermotropism, clonal TCR), lymphomatoid papulosis (CD30+, recurrent, self-healing), APACHE (acral pseudolymphomatous angiokeratoma in children).
Investigations
- Skin biopsy with IHC panel: CD20, CD3, CD4, CD8, CD30, bcl-2, bcl-6, CD10, Ki-67
- Molecular studies: PCR for IgH (B cells) and TCR gene rearrangements — polyclonal = reactive
- Serology: Borrelia antibodies (ELISA/Western blot) in European cases
- PCR for Borrelia DNA from biopsy tissue
Management
- Remove/eliminate the trigger (withdraw offending drug, treat Borrelia infection with doxycycline, remove tattoo pigment)
- Borrelia-associated CLH: Doxycycline 100 mg BD × 3–4 weeks
- Drug-induced: Withdraw causative drug → usually resolves within weeks–months
- Intralesional corticosteroids: For persistent lesions
- Topical/intralesional steroids: Useful for localized disease
- Surgical excision or cryotherapy: For small, persistent lesions
- Hydroxychloroquine or chloroquine: Used in some persistent cases
- Low-dose radiotherapy: Rare, refractory cases
Prognosis: Generally excellent with spontaneous resolution or after removal of the trigger. Long-term follow-up is needed to exclude evolution into true lymphoma, especially in T cell-predominant CLH with clonality.
Source: Dermatology 2-Volume Set 5e (Bolgnia), Ch. 121; Andrews' Diseases of the Skin, p. 2404
b) Rosacea
Definition
Rosacea is a chronic inflammatory disorder primarily affecting the central face, characterized by flushing, persistent erythema, telangiectasia, inflammatory papules and pustules, and phymatous changes. It affects predominantly fair-skinned individuals aged 30–50 years.
Epidemiology
- Affects ~5–10% of the general population
- More common in women (overall), but severe phymatous forms occur almost exclusively in men
- Celtic/northern European ancestry predisposed; less common in darker skin types
- Age of onset: typically 30–50 years
Etiopathogenesis
Multifactorial; exact cause unknown. Key pathogenic factors:
- Vascular dysregulation: Exaggerated neurovascular responses to triggers → flushing → persistent telangiectasia and erythema
- Innate immune dysregulation: Overexpression of cathelicidin (LL-37) processed by KLK5 (kallikrein) → abnormal antimicrobial peptide signaling → inflammation. TLR2 overexpression is also implicated
- Demodex folliculorum: Mite density increased in rosacea; releases bacterial products (Bacillus oleronius) → immune activation
- UV radiation: A major trigger; UV-induced reactive oxygen species → vascular and inflammatory changes
- Neurogenic inflammation: Transient receptor potential (TRP) channels (TRPV1, TRPA1) involved in heat, spice, and UV-induced flushing
- Gastrointestinal association: Small intestinal bacterial overgrowth (SIBO) found in a subset; rifaximin treatment improves rosacea in these patients
Trigger factors: Heat, sunlight (UV), emotional stress, alcohol, hot beverages, spicy foods, exercise, cold/hot weather, topical corticosteroids
Classification (ROSCO/NRS Subtypes)
Old NRS subtype classification (still widely used):
| Subtype | Key Features |
|---|---|
| I — Erythematotelangiectatic (ETR) | Flushing, persistent central facial erythema, telangiectasia; sensitive, easily irritated skin; burning/stinging |
| II — Papulopustular (PPR) | Persistent central facial erythema + inflammatory papules and pinpoint pustules; resembles acne but no comedones |
| III — Phymatous | Skin thickening, nodularity, fibrosis; rhinophyma (nose), gnathophyma (chin), metophyma (forehead), blepharophyma (eyelids); predominantly males |
| IV — Ocular | Blepharitis, conjunctivitis, chalazion, keratitis, iritis; gritty/burning eyes; may precede skin lesions |
Additional variants: Glandular rosacea (edematous papules, sebaceous skin, men > women), granulomatous rosacea (non-caseating granulomas on biopsy), Morbihan disease (solid facial edema)
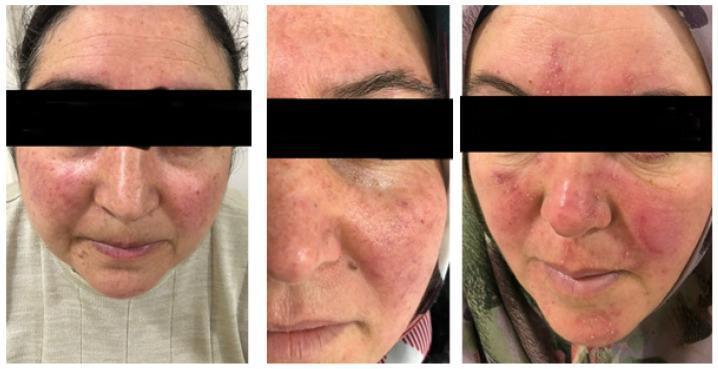
Clinical Features
- Symmetric central facial erythema — cheeks, nose, chin, central forehead; periocular area spared
- Flushing: Prolonged (>10 min), triggered by heat, stress, alcohol; burning/stinging sensation; no sweating or palpitations
- Telangiectasia: Fine surface vessels on cheeks/nasal alae
- Papules and pustules: No comedones (key distinguishing feature from acne)
- Rhinophyma: Bulbous, lobulated thickening of nose — hypertrophy of sebaceous glands + fibrosis
- Ocular rosacea: Blepharitis, recurrent chalazion, keratitis, abnormal Schirmer test (40% of patients)
- Extrafacial: Flushing of neck, chest, ears
Histopathology
- Perivascular and perifollicular lymphohistiocytic infiltrate
- Telangiectatic vessels with solar elastosis
- In granulomatous rosacea: tuberculoid or sarcoidal granulomas
- Demodex mites may be seen in follicles
- No comedones, unlike acne
Differential Diagnosis
- Acne vulgaris (comedones present; younger age)
- Seborrheic dermatitis (greasy scale in nasolabial folds, eyebrows)
- Systemic lupus erythematosus (photosensitive, butterfly rash; ANA positive)
- Dermatomyositis (heliotrope rash; periorbital; proximal weakness)
- Perioral dermatitis (perioral, not central face)
- Carcinoid syndrome (flushing + diarrhea; ↑5-HIAA)
- Polycythemia vera, mastocytosis (no papules)
Management
1. General Measures
- Identify and avoid trigger factors: sun, heat, spicy food, alcohol, hot beverages
- Sunscreen with broad-spectrum SPF ≥30 (zinc oxide/titanium dioxide preferred for sensitive skin)
- Gentle skin care with non-irritating cleansers
2. Topical Therapy
| Agent | Mechanism | Use |
|---|---|---|
| Metronidazole 0.75–1% gel/cream | Anti-inflammatory; anti-Demodex | PPR — maintenance therapy |
| Azelaic acid 15–20% gel/cream | Anti-inflammatory; antikeratinizing | PPR; also helps erythema |
| Ivermectin 1% cream | Anti-Demodex; anti-inflammatory | PPR — superior to metronidazole in trials |
| Brimonidine 0.33% gel (α₂-adrenergic agonist) | Vasoconstriction | ETR — persistent erythema; once morning; lasts ~12 hours |
| Oxymetazoline 1% cream (α₁A-adrenoreceptor agonist) | Vasoconstriction | ETR — persistent erythema |
| Tacrolimus/pimecrolimus | Anti-inflammatory | ETR; also used when stopping long-term topical steroids |
| Topical retinoids (tretinoin) | Normalizes skin | Cautious use; nighttime only |
3. Oral Therapy
| Agent | Dose | Indication |
|---|---|---|
| Doxycycline (subantimicrobial dose) | 40 mg/day modified-release (preferred) OR 50–100 mg BD × 2–3 months | PPR; ocular rosacea |
| Minocycline | 50–100 mg/day | PPR; alternative |
| Isotretinoin | 0.3 mg/kg/day (lower than acne dose) | Severe PPR, granulomatous, rhinophyma; most refractory cases |
| Propranolol / carvedilol | Propranolol 10→40 mg TDS | Flushing reduction |
| Clonidine | 0.05–0.15 mg BD | Flushing (limited efficacy) |
| Rifaximin | For SIBO-associated rosacea | Selected patients |
Note: Systemic antibiotics act primarily via anti-inflammatory (not antibacterial) mechanisms; treatment should be discontinued once inflammatory lesions clear, with topical agents used for maintenance.
4. Surgical & Physical Treatments
| Modality | Indication |
|---|---|
| Pulsed Dye Laser (PDL) / KTP laser | Telangiectasia and persistent erythema |
| Intense Pulsed Light (IPL) | Erythema and telangiectasia; comparable to PDL |
| CO₂ laser / dermabrasion | Rhinophyma reshaping |
| Electrocautery / tangential excision | Rhinophyma |
| Cyclosporine ophthalmic emulsion | Ocular rosacea |
5. Management by Subtype
ETR: Trigger avoidance + sunscreen → Brimonidine/Oxymetazoline gel (erythema)
→ IPL/PDL (telangiectasia)
PPR: Topical metronidazole / azelaic acid / ivermectin
→ Add oral doxycycline 40 mg for moderate-severe disease
→ Switch to isotretinoin for refractory cases
Phymatous: Isotretinoin (early) + CO₂ laser / surgery (advanced rhinophyma)
Ocular: Oral doxycycline + warm compresses + cyclosporine eye drops
Prognosis
Rosacea is a chronic, relapsing disorder with no permanent cure. However, with appropriate trigger avoidance and treatment, symptoms can be controlled in most patients. Rhinophyma and fibrotic changes, once established, do not regress with medical therapy alone. Ocular rosacea, if untreated, can lead to corneal scarring and visual impairment.
Sources: Andrews' Diseases of the Skin, p. 4195; Fitzpatrick's Dermatology Vol 1–2, p. 1449; Dermatology 2-Volume Set 5e (Bolgnia)
Write briefly about-a)lipodermatosclerosis and b) Sezary syndrome in 10 marks format for 3rd yr dermatology post graduate theory examination
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.