Paracetamol
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Searching PubMed
paracetamol mechanism of action
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive answer on paracetamol's mechanism and pharmacology.
Paracetamol (Acetaminophen) — Mechanism of Action & Pharmacology
Chemical Identity
Paracetamol (acetaminophen; N-acetyl-p-aminophenol) is a para-aminophenol derivative. First synthesised in 1878, it became the dominant analgesic in the 1950s after phenacetin was withdrawn for renal toxicity. It is now the most widely used drug worldwide and forms Step 1 of the WHO analgesic ladder.
Mechanisms of Action
The mechanism is multifactorial and not fully resolved, involving both central and peripheral components.
1. COX Inhibition — Central, Not Peripheral
Paracetamol is a non-selective inhibitor of cyclooxygenase (COX-1 and COX-2), but critically its inhibition is predominantly central with minimal peripheral effect. This explains why it shares the analgesic and antipyretic effects of NSAIDs but lacks meaningful anti-inflammatory activity.
The mechanism of COX inhibition is distinct from NSAIDs:
- NSAIDs compete directly with arachidonic acid at the COX active site.
- Paracetamol does not enter the active site of the COX enzyme and does not bind to the COX catalytic site.
- Instead, it reduces the haem at the peroxidase site of COX, thereby preventing enzyme activation.
This peroxidase-site interaction is concentration-dependent and is overcome in peripheral inflamed tissues, which have high peroxide concentrations — explaining the drug's lack of anti-inflammatory efficacy at those sites. In the CNS (low peroxide environment), inhibition is effective.
"The drug is primarily a centrally acting inhibitor of the COX enzyme with minimal peripheral effects. Acetaminophen neither enters the active site of the COX enzyme nor binds to the COX site. Instead, it prevents COX activation by reducing heme at the peroxidase site of the enzyme." — Barash, Cullen & Stoelting's Clinical Anesthesia, 9e
2. Descending Serotoninergic Pathways
Paracetamol may modulate descending inhibitory serotoninergic (5-HT) pathways from the brain to the spinal cord. Activation of these pathways suppresses ascending nociceptive signals. This central component contributes meaningfully to analgesia.
3. Endocannabinoid System
Paracetamol is deacylated in the brain to p-aminophenol, which then conjugates with arachidonic acid to form AM404, an endocannabinoid analogue. AM404 activates CB₁ cannabinoid receptors and inhibits endocannabinoid reuptake — this is now thought to be a significant pathway.
4. Additional Receptor Interactions
Evidence supports modulation of:
- TRPV1 (transient receptor potential vanilloid 1) channels — desensitisation reduces nociceptor activity
- Opioid receptors — indirect modulation
- NMDA receptors — attenuating central sensitisation
- Nitric oxide (NO) synthesis — paracetamol inhibits NO synthesis in the spinal cord, dampening central sensitisation
Pharmacokinetics
| Parameter | Detail |
|---|---|
| Onset | Oral/rectal: ~40 min; peak effect ~1 hour |
| Oral bioavailability | 63–89% (variable) |
| Rectal bioavailability | 24–98% (highly variable) |
| IV bioavailability | 100% (overcomes absorption variability) |
| Duration of action | 4–6 hours |
| Protein binding | Low at therapeutic doses |
| Distribution | Crosses blood-brain barrier (essential for central action) |
| CYP interaction | Substrate of CYP3A4; does not inhibit fentanyl metabolism at therapeutic doses |
Metabolism (Hepatic)
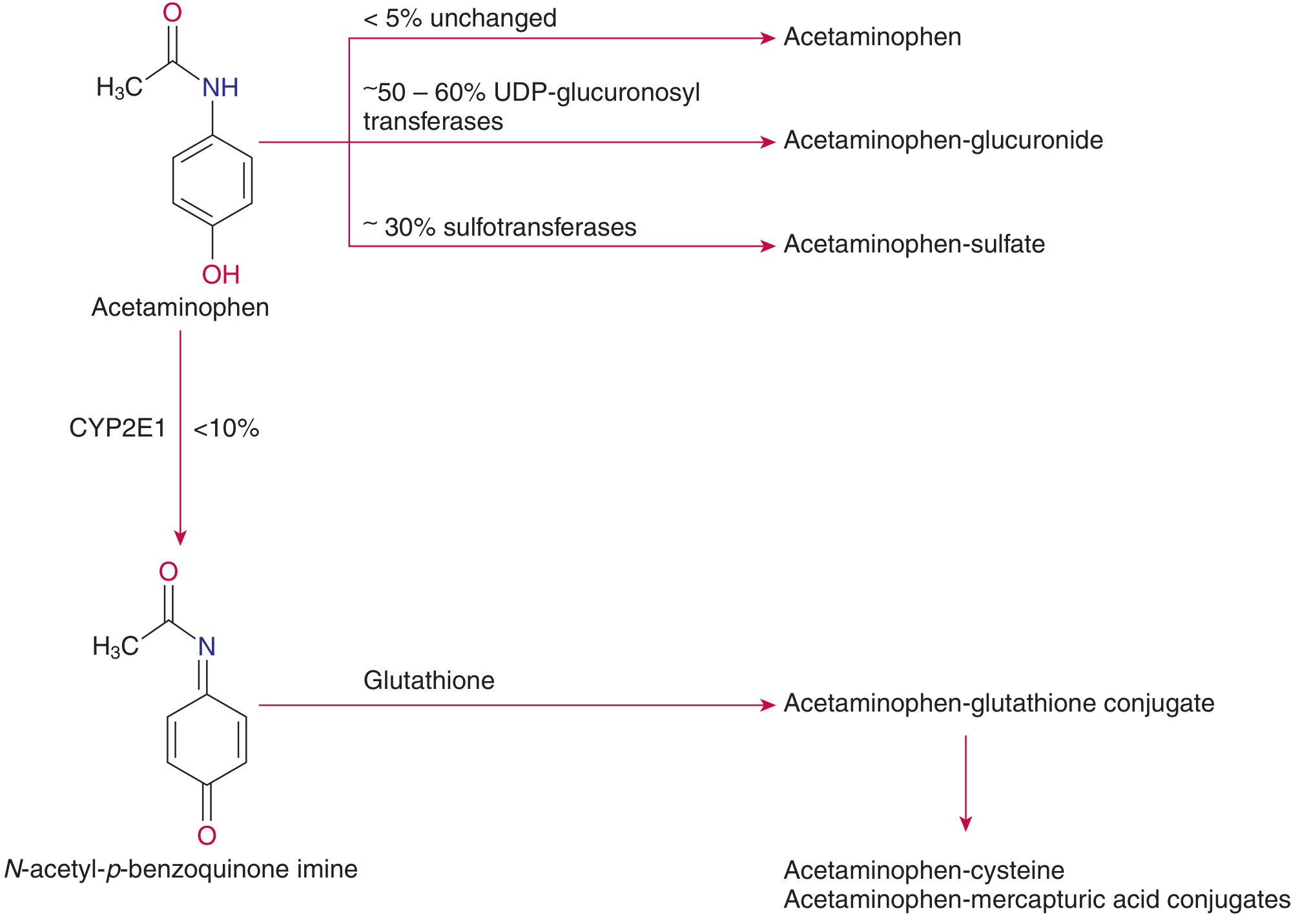
Pathways of acetaminophen metabolism — Tietz Textbook of Laboratory Medicine, 7e
At therapeutic doses, three pathways operate in the liver:
- ~50–60% → glucuronide conjugate (via UDP-glucuronosyltransferases) — non-toxic, renally excreted
- ~30% → sulphate conjugate (via sulfotransferases) — non-toxic, renally excreted
- <10% → oxidised via CYP2E1 (and CYP3A4) to the reactive toxic intermediate NAPQI (N-acetyl-p-benzoquinone imine), which is then rapidly detoxified by conjugation with glutathione → mercapturic acid conjugate
In overdose: the sulphation pathway saturates, diverting more paracetamol to CYP2E1. Glutathione stores become depleted, NAPQI accumulates, and it arylates hepatocellular proteins → centrilobular hepatic necrosis.
Key Pharmacological Properties
| Property | Detail |
|---|---|
| Analgesic | Yes — mild-to-moderate pain |
| Antipyretic | Yes (central hypothalamic COX inhibition → reduced PGE₂) |
| Anti-inflammatory | Weak / negligible |
| GI effects | Minimal — does not cause GI ulceration or bleeding |
| Platelet function | No effect (unlike NSAIDs) |
| Cardiorenal effects | None at therapeutic doses |
| Opioid-sparing | Yes — 20–30% reduction in opioid requirements as part of multimodal analgesia |
| Bone fusion | Does not impair, unlike NSAIDs |
Summary
Paracetamol's analgesia and antipyresis arise primarily from central COX inhibition (via the peroxidase site, not the arachidonic acid binding site), supplemented by serotoninergic descending pathway modulation, endocannabinoid (AM404) activity, and TRPV1/NMDA/NO pathway effects. Its negligible peripheral COX inhibition explains the absence of anti-inflammatory effects and the favourable GI/platelet/renal safety profile that distinguishes it from classical NSAIDs.
Sources: Barash, Cullen & Stoelting's Clinical Anesthesia 9e | Tietz Textbook of Laboratory Medicine 7e | Goodman & Gilman's Pharmacological Basis of Therapeutics | Bailey & Love's Short Practice of Surgery 28e | Miller's Anesthesia 10e
This is a shared conversation. Sign in to Orris to start your own chat.