Restrictive respiratory diseases
Reading File
Finding Sources
Finding Sources
Searching PubMed
"restrictive lung disease"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed for a comprehensive answer. Here is a complete overview of restrictive respiratory diseases:
Restrictive Respiratory Diseases
Definition & Physiology
Restrictive lung diseases are characterized by a reduced total lung capacity (TLC) — the lung cannot fully expand. On pulmonary function testing (PFT):
- ↓ TLC, ↓ FVC, ↓ FEV1 — all volumes reduced
- FEV1/FVC ratio is normal or elevated (≥0.70) — distinguishing feature from obstructive disease
- ↓ DLCO — commonly reduced in parenchymal disease
Causes are divided into intrinsic (lung parenchyma/interstitium) and extrinsic (chest wall, pleura, neuromuscular) categories.
Classification
I. Intrinsic (Parenchymal) — Interstitial Lung Diseases (ILDs)
Diffuse parenchymal lung diseases represent >200 heterogeneous conditions affecting the lung interstitium (the space between the alveolar epithelium and endothelium) with varying degrees of inflammation and fibrosis. Most patients present with progressive exertional dyspnea or persistent dry cough.
A. Idiopathic Interstitial Pneumonias (IIPs)
1. Idiopathic Pulmonary Fibrosis (IPF)
- Most common ILD of unknown cause; prevalence 50–200 per 100,000
- Diagnosed in the 5th–6th decade; more common in men; associated with smoking
- 50% 3–5 year survival — poor prognosis
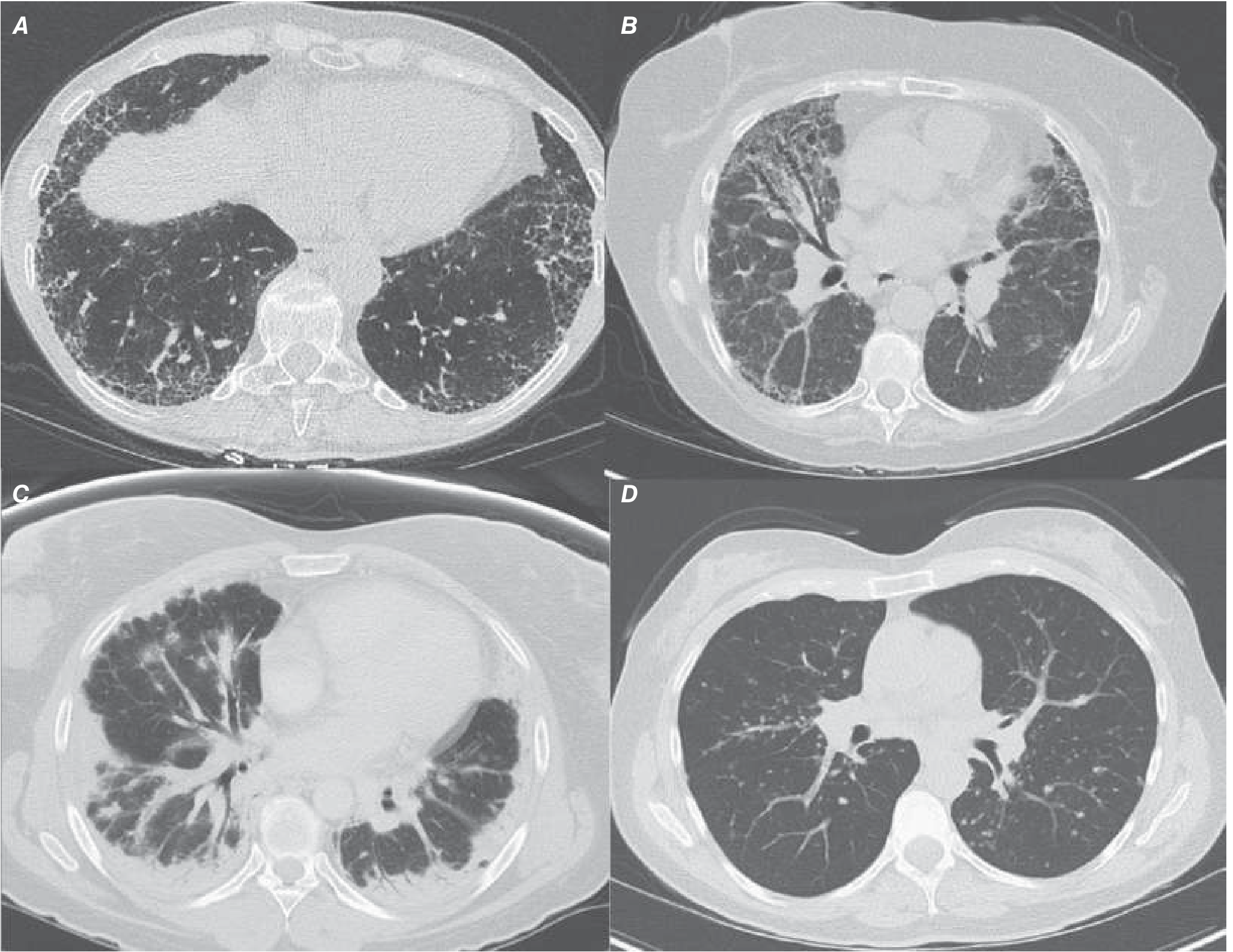
- HRCT: Bilateral subpleural reticular markings with posterior/basal predominance; honeycombing and traction bronchiectasis → "UIP pattern"
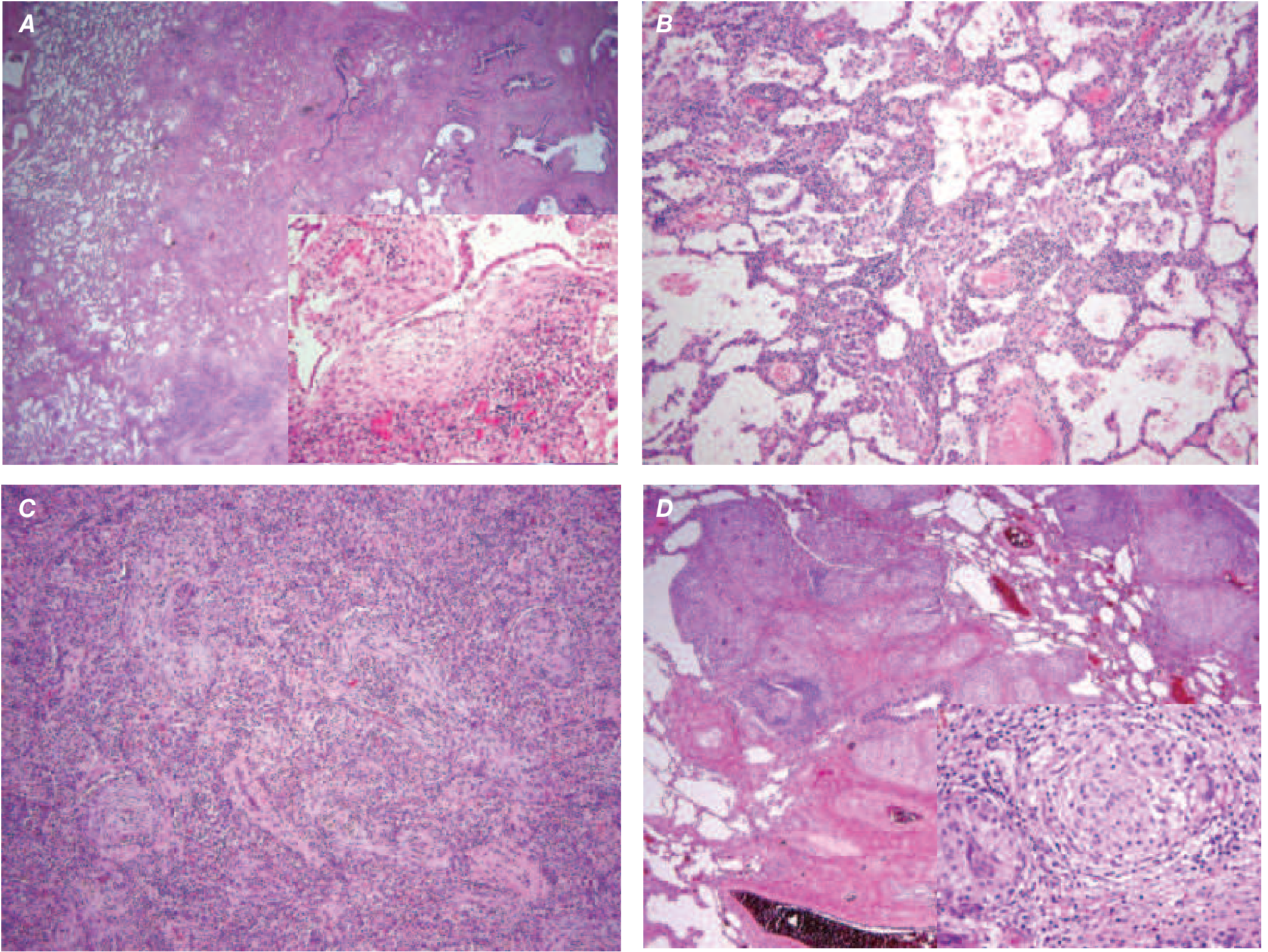
- Histology: Subpleural reticulation + honeycomb changes + fibroblast foci (subepithelial myofibroblasts/collagen); temporal and spatial heterogeneity → Usual Interstitial Pneumonia (UIP)
- Treatment: Antifibrotics — pirfenidone and nintedanib (proven in 2014 trials to slow FVC decline); lung transplantation for eligible patients; O₂ therapy + pulmonary rehab
2. Nonspecific Interstitial Pneumonia (NSIP)
- More common in nonsmoking females in the 5th decade; frequently associated with CTD
- 5-year survival >80% — better prognosis than IPF
- HRCT: Bilateral, symmetric subpleural ground-glass and reticular opacities; lower lung zone predominance; subpleural sparing (unlike IPF); no or rare honeycombing
- Histology: Uniform interstitial inflammation and fibrosis (cellular > fibrotic in prognosis); absence of honeycomb change; fibroblast foci rare
- Treatment: Oral steroids (prednisone), cytotoxic agents (mycophenolate, azathioprine, cyclophosphamide), biologics (rituximab, tocilizumab); antifibrotics if progressive
3. Cryptogenic Organizing Pneumonia (COP)
- HRCT: Patchy, sometimes migratory, subpleural consolidative opacities ± ground-glass; reversed halo/atoll sign
- Histology: Patchy plugs of granulation tissue in small airways, alveolar ducts, and alveoli; mononuclear infiltration; foamy macrophages
4. Smoking-Related ILDs
- RB-ILD (Respiratory Bronchiolitis-ILD) and DIP (Desquamative Interstitial Pneumonia): strong association with tobacco smoke
- Pulmonary Langerhans Cell Histiocytosis (PLCH) also in this category
5. Acute Exacerbations of IIPs
- Can occur in any ILD with pulmonary fibrosis; most severe in IPF
- Acute onset (<30 days) of respiratory distress/hypoxemia on background of pulmonary fibrosis
- Mortality >85%; histology shows DAD pattern
- Treatment is largely supportive; nintedanib may reduce AE rate in IPF
B. ILD Associated with Connective Tissue Disease (CTD-ILD)
| CTD | Key Features |
|---|---|
| Systemic Sclerosis (SSc) | Most common CTD-ILD; ~50% in diffuse SSc, ~30% in limited; NSIP pattern most common; esophageal dysmotility contributes via aspiration. Treatment: mycophenolate (preferred over cyclophosphamide), tocilizumab, nintedanib |
| Rheumatoid Arthritis (RA) | More common in males with RA; UIP most common pattern; progressive fibrosis responds to antifibrotics |
| Dermatomyositis/Polymyositis | ILD in up to 45% with anti-synthetase antibodies (anti-Jo-1); NSIP/COP patterns; anti-synthetase syndrome |
| SLE / Sjögren's | Less frequent ILD involvement |
C. Granulomatous ILDs
Sarcoidosis
- Can be asymptomatic or present with dyspnea, cough, fatigue, eye/skin/joint findings
- HRCT: Mediastinal and hilar lymphadenopathy + circular-nodular opacities along bronchovascular bundles (classic); also miliary nodules, ground-glass, or mosaic attenuation
- Histology: Non-caseating granulomas (hallmark)
- Restrictive pattern on PFTs (can also be obstructive or mixed)
Hypersensitivity Pneumonitis (Extrinsic Allergic Alveolitis)
- Caused by repeated inhalation of organic antigens (farmer's lung, bird fancier's lung)
- Acute, subacute, and chronic forms; chronic form leads to progressive fibrosis
- HRCT: Upper lobe predominant fibrosis, air trapping, mosaic pattern
D. Occupational/Environmental ILDs (Pneumoconioses)
- Silicosis — crystalline silica exposure; upper lobe nodules, eggshell calcification of hilar nodes
- Asbestosis — lower lobe fibrosis, pleural plaques/mesothelioma risk
- Coal Worker's Pneumoconiosis — progressive massive fibrosis
- Berylliosis — resembles sarcoidosis
II. Extrinsic Causes
| Category | Examples |
|---|---|
| Chest wall deformities | Kyphoscoliosis, pectus excavatum (severe) — prone to desaturation, especially at altitude |
| Neuromuscular disease | ALS, muscular dystrophy, Guillain-Barré, diaphragm paralysis — reduced inspiratory effort |
| Pleural disease | Large pleural effusion, fibrothorax, mesothelioma |
| Obesity | Reduces FRC and ERV (obesity hypoventilation syndrome) |
| Abdominal distension | Ascites, pregnancy — elevated diaphragm |
Key Diagnostic Approach
PFTs
- Restriction: TLC <80% predicted; FEV1/FVC ≥0.70
- Severity graded by FVC: Mild ≥70%, Moderate 50–69%, Severe <50%
- DLCO reduction suggests parenchymal disease (ILD) vs. preserved DLCO in pure chest wall restriction
HRCT Patterns (Figure 304-2 from Harrison's)
Histopathology (Figure 304-3 from Harrison's)
Diagnostic Workup
- History: Exposures (occupational, birds, hay), smoking, medications, CTD symptoms
- Serology: ANA, anti-dsDNA, Scl-70 (anti-topo I), RF, anti-CCP, anti-Jo-1, anti-synthetase panel
- HRCT chest — often definitive; multidisciplinary discussion (MDD) approach
- BAL — cellular differential (neutrophilia in IPF, lymphocytosis in HP/sarcoidosis)
- Lung biopsy — VATS surgical biopsy when CT non-diagnostic; transbronchial biopsy or cryo-biopsy for specific conditions
Treatment Summary
| Disease | First-Line | Second-Line |
|---|---|---|
| IPF | Nintedanib or Pirfenidone | Lung transplant |
| CTD-ILD (SSc) | Mycophenolate, Tocilizumab, Nintedanib | Cyclophosphamide, transplant |
| CTD-ILD (RA) | Immunosuppressives ± antifibrotics | Lung transplant |
| DM/PM-ILD | Prednisone + mycophenolate/azathioprine | Rituximab, transplant |
| COP/NSIP | Prednisone | Cytotoxics, biologics |
| Sarcoidosis | Oral corticosteroids | Methotrexate, hydroxychloroquine |
| HP (chronic) | Antigen avoidance + steroids | Antifibrotics if progressive |
| All progressive ILD | Pulmonary rehab, supplemental O₂ | Lung transplantation |
Sources: Harrison's Principles of Internal Medicine 22E (2025), Chapter 304 (Interstitial Lung Disease) | Murray & Nadel's Textbook of Respiratory Medicine | Fishman's Pulmonary Diseases and Disorders
This is a shared conversation. Sign in to Orris to start your own chat.