Bleeding disorders pathology
Reading File
Finding Sources
Finding Sources
Searching PubMed
"bleeding disorders" pathology hemostasis
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Searching Images
hemophilia bleeding disorder coagulation factor deficiency pathology
I now have all the content needed for a comprehensive answer. Let me compile it.
Bleeding Disorders — Pathology
Source: Robbins & Kumar Basic Pathology, 10th ed.
Overview & Classification
Bleeding disorders are characterized by abnormal bleeding that may be spontaneous or provoked by trauma/surgery. They arise from defects in one or more components of hemostasis:
| Category | Examples |
|---|---|
| Vascular fragility | Scurvy, amyloidosis, glucocorticoid excess, vasculitis |
| Platelet disorders | Thrombocytopenia, qualitative platelet defects |
| Coagulation factor deficiencies | Hemophilia A & B, von Willebrand disease |
| Mixed / consumptive | DIC |
Diagnostic Laboratory Tests
| Test | Pathway Assessed | Prolonged By |
|---|---|---|
| PT (Prothrombin Time) | Extrinsic + common pathways | Deficiency of factors V, VII, X, prothrombin, or fibrinogen; acquired inhibitors |
| PTT (Partial Thromboplastin Time) | Intrinsic + common pathways | Deficiency of factors V, VIII, IX, X, XI, XII, prothrombin, or fibrinogen |
| Platelet count | Quantitative platelet assessment | Reference: 150,000–450,000/μL |
| Platelet function tests | Aggregation studies, vWF assays | Qualitative defects, vWF deficiency |
| Specific factor assays | Individual factor levels | Factor-specific deficiencies |
1. Vascular Fragility Disorders
Bleeding due purely to vascular wall weakness presents with:
- "Spontaneous" petechiae and ecchymoses in skin and mucous membranes (likely from trivial trauma)
- Normal coagulation tests (PT, PTT, platelet count)
Causes include:
- Scurvy — vitamin C deficiency impairs collagen synthesis
- Amyloidosis — amyloid deposits in vessel walls
- Chronic glucocorticoid use — atrophies perivascular connective tissue
- Hereditary connective tissue disorders (e.g., Ehlers-Danlos)
- Infectious/hypersensitivity vasculitides
2. Thrombocytopenia
Defined as platelet count <150,000/μL. Clinically significant spontaneous bleeding rarely occurs until counts fall below ~20,000/μL. Causes include:
- Decreased production: bone marrow failure, aplastic anemia, infiltrative neoplasms
- Increased destruction:
- Immune thrombocytopenic purpura (ITP) — autoantibodies (usually IgG) against platelet membrane glycoproteins (GPIIb/IIIa); splenic destruction
- Drug-induced (heparin-induced thrombocytopenia, HIT)
- TTP/HUS — microangiopathic consumption
- Sequestration: hypersplenism
- Dilutional: massive transfusion
Qualitative platelet defects (normal count but abnormal function):
- Uremia (acquired)
- Glanzmann thrombasthenia — absent GPIIb/IIIa (inherited)
- Bernard-Soulier syndrome — absent GPIb (collagen receptor)
- Aspirin/NSAIDs — irreversible COX inhibition
3. Coagulation Factor Deficiencies
Von Willebrand Disease (vWD)
The most common inherited bleeding disorder. vWF has two critical roles:
- Mediates platelet adhesion to subendothelial collagen
- Carries factor VIII in plasma (protects it from proteolysis)
| Type | Defect | Frequency |
|---|---|---|
| Type 1 | Quantitative reduction (partial) | ~75% of cases |
| Type 2 | Qualitative dysfunction | Variable subtypes |
| Type 3 | Complete absence (severe) | Rare; AR |
Lab findings: Prolonged bleeding time, prolonged PTT (due to low factor VIII), normal PT. Ristocetin cofactor assay is reduced.
Hemophilia A: Factor VIII Deficiency
- X-linked recessive (males affected; females carriers)
- Accounts for ~80% of hemophilias
- Factor VIII is a cofactor for factor IXa in the intrinsic pathway
Clinical features (severity depends on factor VIII level):
- Severe (<1% factor VIII): spontaneous hemarthroses, deep muscle hematomas
- Moderate (1–5%): bleeding after mild trauma
- Mild (5–30%): bleeding only after significant trauma/surgery
Hallmarks:
- Hemarthrosis (bleeding into joints) → most common; leads to chronic joint deformity
- Deep tissue hematomas
- Post-circumcision / post-procedure bleeding
- Petechiae and superficial bruising are not features (contrast with platelet disorders)
Lab: Prolonged PTT, normal PT, normal platelet count. Specific factor VIII assay confirms diagnosis.
Treatment: Recombinant factor VIII infusion. A bispecific antibody (emicizumab) that bridges factor IX and factor X bypasses the need for factor VIII entirely — particularly useful in patients who develop anti-factor VIII inhibitor antibodies (~15% of severe HA).
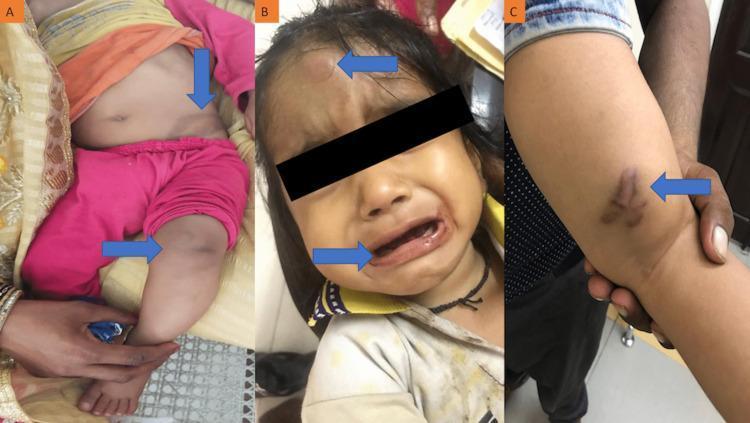
Hemophilia B: Factor IX Deficiency
- X-linked recessive, clinically indistinguishable from hemophilia A
- Much less common (~20% of hemophilias)
- Lab: Prolonged PTT, normal PT
- Treatment: Recombinant factor IX infusion
4. Disseminated Intravascular Coagulation (DIC)
Probably causes more bleeding than all congenital coagulation disorders combined, as it complicates many acute illnesses.
Pathogenesis
DIC is triggered by two main mechanisms:
-
Release of tissue factor/procoagulants into circulation:
- Obstetric emergencies (amniotic fluid, placental tissue)
- Cancer cells (esp. acute promyelocytic leukemia, adenocarcinomas)
- Bacterial endotoxins → monocyte IL-1/TNF → upregulate tissue factor on endothelium + downregulate thrombomodulin (→ less protein C activation)
-
Widespread endothelial injury:
- Antigen-antibody complex deposition (SLE)
- Temperature extremes (heat stroke, burns)
- Infections (meningococcemia, rickettsiae)
- Sepsis
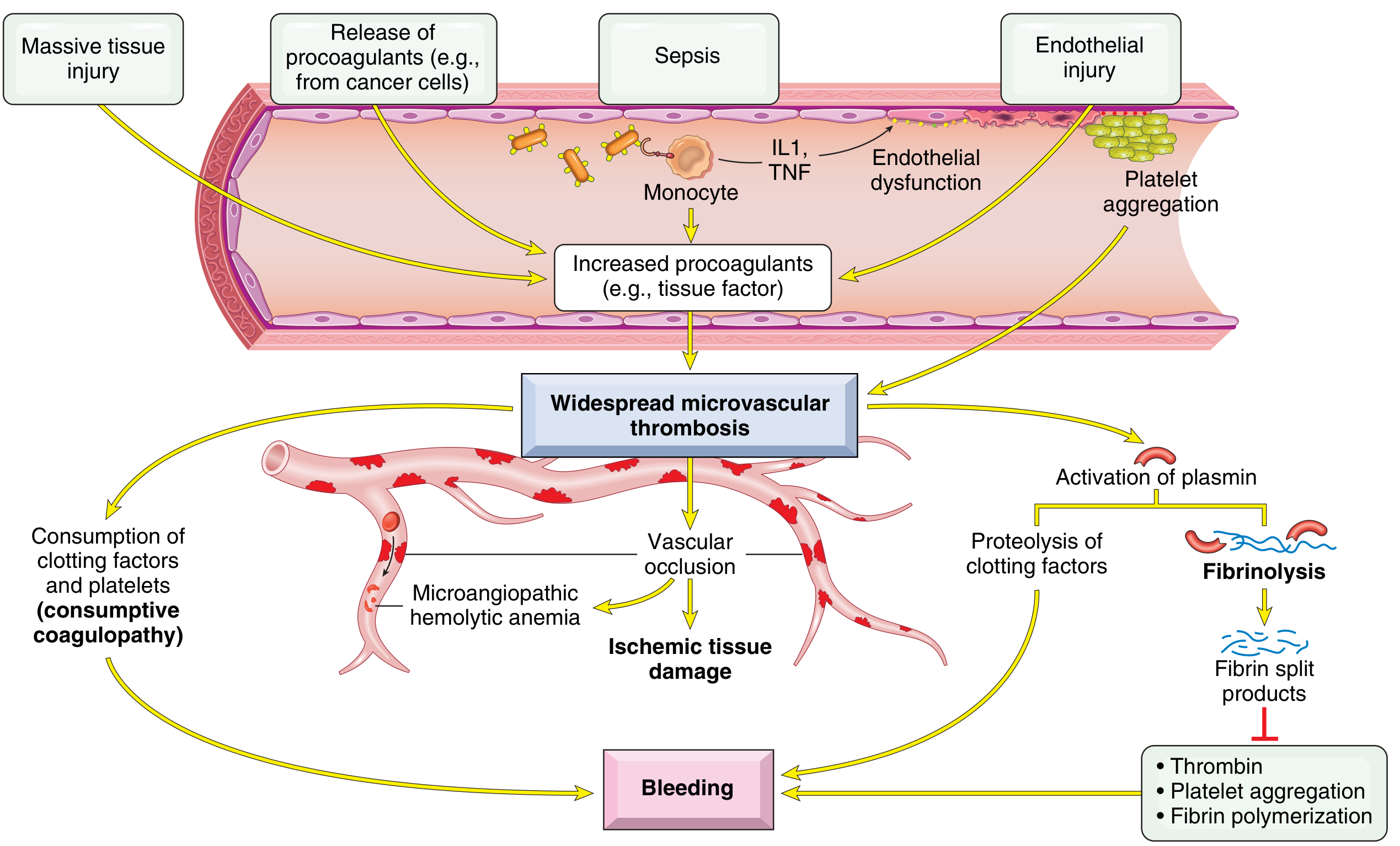
Major DIC Triggers (Table 10.10)
| Category | Examples |
|---|---|
| Obstetric | Abruptio placentae, retained nonviable fetus, amniotic fluid embolism, eclampsia |
| Infections | Gram-negative/positive sepsis, meningococcemia, Rocky Mountain spotted fever, malaria |
| Neoplasms | Pancreatic/prostatic/lung carcinoma, acute promyelocytic leukemia |
| Massive tissue injury | Trauma, burns, extensive surgery (especially brain trauma) |
| Miscellaneous | Intravascular hemolysis, snakebite, giant hemangioma, shock, liver disease |
Consequences of DIC (Dual Paradox)
1. Thrombotic phase → widespread microvascular fibrin thrombi:
- Ischemia and microinfarcts (kidneys, adrenals, brain, heart)
- Microangiopathic hemolytic anemia — RBCs sheared by fibrin strands
2. Hemorrhagic phase (consumptive coagulopathy):
- Platelets and clotting factors consumed faster than they are replenished
- Secondary fibrinolysis activated → plasmin cleaves fibrin AND factors V and VIII
- Fibrin degradation products (FDPs) inhibit platelet aggregation, have anti-thrombin activity, and impair fibrin polymerization → compounding hemorrhage
Morphology in DIC
Microthrombi are found in arterioles and capillaries of:
- Kidneys — glomerular fibrin thrombi → focal glomerulitis → cortical microinfarcts → bilateral renal cortical necrosis (severe)
- Adrenals → Waterhouse-Friderichsen syndrome
- Brain → microinfarcts + foci of hemorrhage
- Lungs, heart, liver also affected
Lab Findings in DIC
| Test | Finding |
|---|---|
| PT & PTT | Both prolonged |
| Platelet count | Low (consumptive) |
| Fibrinogen | Decreased |
| D-dimer / FDPs | Elevated (fibrinolysis) |
| Peripheral smear | Schistocytes (microangiopathic hemolysis) |
Summary: Distinguishing Patterns
| Feature | Vascular | Platelet Defect | Coagulation Factor Defect | DIC |
|---|---|---|---|---|
| Petechiae | + | ++ | − | ± |
| Hemarthrosis | − | − | ++ | − |
| PT | N | N | ↑ (factor VII, X, V, II) | ↑ |
| PTT | N | N | ↑ (VIII, IX, XI, XII) | ↑ |
| Platelet count | N | ↓ or N | N | ↓ |
| Bleeding time | ↑ | ↑ | N | ↑ |
Robbins & Kumar Basic Pathology, 10th ed., pp. 426–431
This is a shared conversation. Sign in to Orris to start your own chat.