how to write case history (and how to rule out other diagnosis from history) with clinical examination(and how to rule out other diagnosis from clinical examination) of a patient with peripheral neuropathy
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
I now have a comprehensive set of source material. Let me compile the full, detailed answer.
Case History and Clinical Examination in Peripheral Neuropathy
Sources: Harrison's Principles of Internal Medicine 22E (2025); Adams & Victor's Principles of Neurology, 12th Ed; Goldman-Cecil Medicine
Diagnostic Approach Overview
The clinician facing a patient with suspected peripheral neuropathy must answer four sequential questions (Adams & Victor):
- Is this truly a peripheral nervous system disease, or a CNS/neuromuscular junction/muscle disorder?
- What is the topographic pattern (polyneuropathy, mononeuropathy, radiculopathy, plexopathy)?
- Is the process motor, sensory, autonomic, or mixed - and is it axonal or demyelinating?
- Is the neuropathy acquired or hereditary?
Harrison's frames this as three main goals: localize the lesion, identify the cause, determine treatment - approached through 7 Key Questions answered from history and examination.
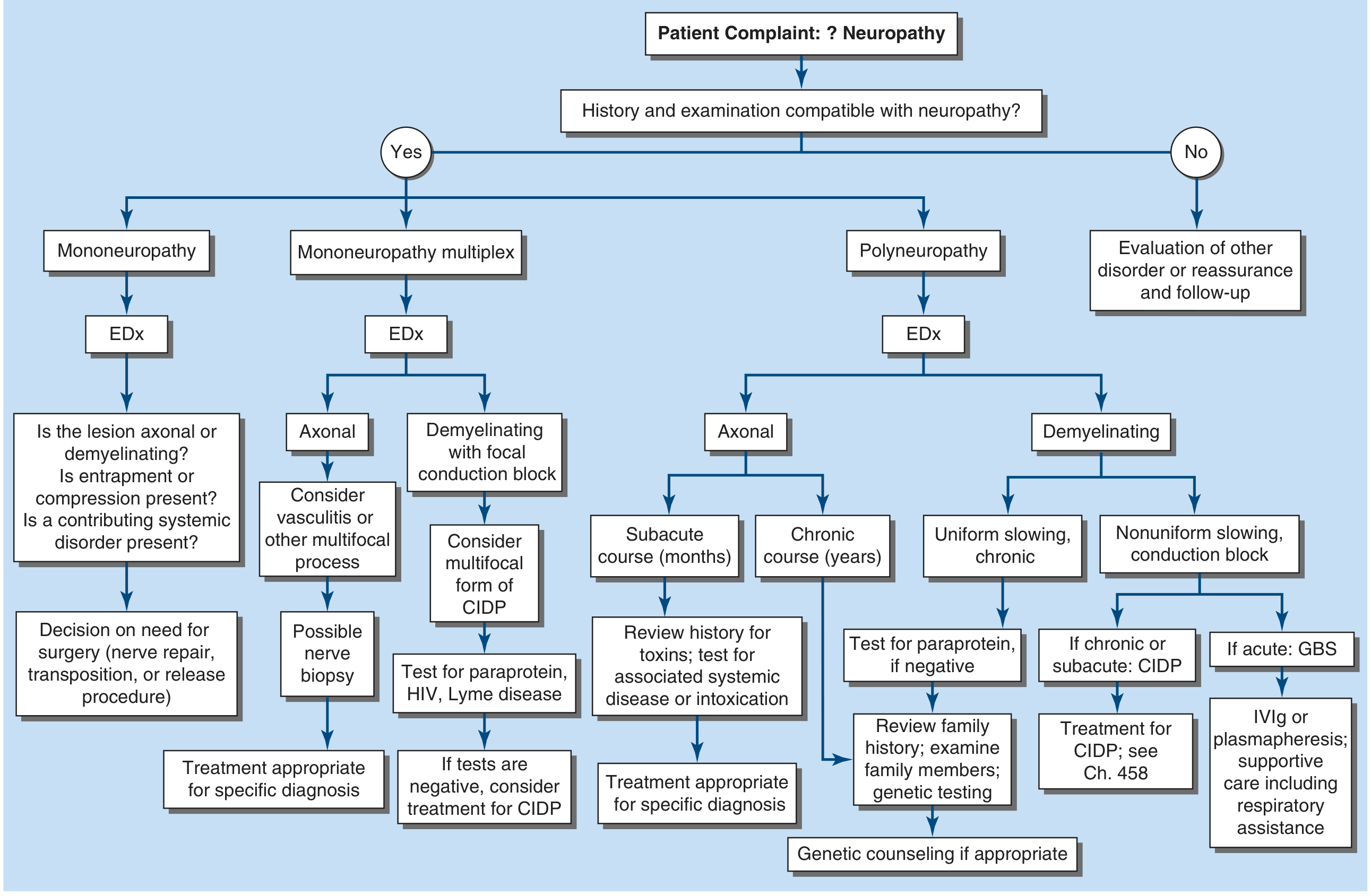
Figure: Approach to the evaluation of peripheral neuropathies (Harrison's 22E)
PART I: CASE HISTORY
A. Chief Complaint and Onset
Begin by documenting what symptoms brought the patient in - sensory symptoms (numbness, tingling, burning, pain), motor symptoms (weakness, tripping, difficulty with fine tasks), or autonomic symptoms (dizziness on standing, sweating abnormalities, bladder/bowel/sexual dysfunction).
Ask specifically:
- Onset: sudden vs. gradual
- Duration and progression: acute (days-4 weeks), subacute (4-8 weeks), chronic (>8 weeks)
- Course: monophasic, progressive, relapsing-remitting
- Distribution: feet first? Both feet simultaneously? One limb? Multiple sites?
A chronic, slowly progressive symmetrical sensory-predominant course starting in the feet is the classic pattern of a length-dependent polyneuropathy (e.g., diabetic, toxic, idiopathic).
An acute onset (days to 4 weeks) of ascending weakness with areflexia strongly suggests Guillain-Barré syndrome (GBS).
An asymmetric, multifocal or episodic onset suggests mononeuropathy multiplex (vasculitis, diabetes, sarcoid, leprosy).
B. The 7 Key Questions from History (Harrison's)
Question 1: What Systems Are Involved?
Ask about:
- Motor: weakness, foot drop, difficulty with grip, trips/falls
- Sensory: numbness, tingling, burning, "pins and needles," pain, loss of balance in the dark
- Autonomic: fainting, orthostatic dizziness, heat intolerance, anhidrosis, bowel/bladder/sexual dysfunction
How this rules out other diagnoses:
- Pure motor involvement without sensory symptoms → consider motor neuron disease (ALS), myopathy, or neuromuscular junction disorder (myasthenia gravis, Lambert-Eaton) rather than polyneuropathy
- Prominent autonomic dysfunction without diabetes → strongly suggests amyloid polyneuropathy or hereditary sensory and autonomic neuropathy (HSAN)
- Sensory-only with normal strength → small-fiber neuropathy (DM, glucose intolerance, amyloid) or sensory neuronopathy/ganglionopathy (paraneoplastic, Sjögren's)
Question 2: What Is the Distribution of Weakness?
- Distal-only weakness: typical axonal polyneuropathy (DM, toxic, idiopathic)
- Proximal AND distal symmetric weakness: hallmark of acquired immune demyelinating polyneuropathies - GBS (acute) and CIDP (chronic); this pattern cannot be overemphasized as it identifies a treatable disorder
- Asymmetric/multifocal weakness: radiculopathy, plexopathy, compressive mononeuropathy, or mononeuropathy multiplex (vasculitis, leprosy, diabetes)
- Unilateral extremity weakness without sensory symptoms: consider ALS (motor neuron disease) vs. multifocal motor neuropathy (MMN) - the latter is treatable with IVIg
Rules out:
- Proximal-predominant weakness without sensory loss → myopathy or NMJ disorder, not polyneuropathy
- Neck extensor/bulbar weakness → ALS, isolated bulbar GBS, Kennedy's syndrome
Question 3: What Is the Nature of Sensory Involvement?
- Pain, burning, temperature loss, allodynia = small-fiber (C and A-delta fibers) involvement → DM, glucose intolerance, amyloid, HIV, Fabry's disease
- Vibration and proprioception loss (with normal pain/temperature) = large-fiber involvement → vitamin B12 deficiency, CIDP, hereditary neuropathy (CMT)
- Severe proprioceptive loss with imbalance, positive Romberg, pseudoathetosis = sensory neuronopathy/ganglionopathy → paraneoplastic (anti-Hu), Sjögren's syndrome, cisplatin toxicity, vitamin B6 toxicity, CANVAS syndrome
Rules out:
- Loss of vibration/proprioception with hyperreflexia and UMN signs → posterior column + corticospinal lesion (subacute combined degeneration from B12 deficiency, MS, cervical myelopathy) - NOT peripheral neuropathy alone
- Pain and temperature loss with preserved vibration/proprioception = dorsal horn or spinothalamic tract lesion (syringomyelia, Brown-Séquard) rather than peripheral neuropathy
Question 4: Is There Upper Motor Neuron Involvement?
Always ask about: spasticity, brisk reflexes, upgoing plantars (Babinski), clonus, sphincter disturbance
- Peripheral neuropathy produces lower motor neuron signs: weakness, wasting, hyporeflexia/areflexia, sensory loss
- If UMN + LMN signs coexist: suspect ALS (UMN + LMN), vitamin B12 deficiency (combined system degeneration), adrenomyeloneuropathy, or hereditary spastic paraplegia with neuropathy (HSP-plus)
- UMN signs alone with no sensory loss → motor neuron disease (ALS, PLS), NOT peripheral neuropathy
Question 5: What Is the Temporal Evolution?
| Course | Duration | Suggested Cause |
|---|---|---|
| Acute | Days-4 weeks | GBS, porphyria, diphtheria, toxic (arsenic, thallium) |
| Subacute | 4-8 weeks | CIDP, deficiency states (B1, B12), drug toxicity, vasculitic neuropathy, paraneoplastic |
| Chronic progressive | >8 weeks-years | Diabetic, hereditary (CMT), CIDP, paraproteinemic, amyloid |
| Relapsing-remitting | Episodic | CIDP, hereditary neuropathy with pressure palsies (HNPP), porphyria |
Rules out:
- Hyperacute onset (hours-1 day) of profound weakness → consider acute ischemic neuropathy, porphyria, or critical illness polyneuropathy
- Very slow insidious course over decades, often with pes cavus and no sensory symptoms → hereditary neuropathy (CMT) rather than acquired
Question 6: Is There Evidence of a Hereditary Neuropathy?
Ask specifically:
- Family history of neuropathy, gait difficulty, foot deformity, inability to run since childhood
- Pes cavus (high arched feet) and hammer toes (especially if present since childhood)
- Lack of sensory symptoms despite sensory signs on examination (highly characteristic of CMT - patients adapt to slow fiber loss)
- Scoliosis, kyphoscoliosis
Hereditary clues in history:
- CMT: onset in childhood/teens, slowly progressive, family members similarly affected, no sensory complaints despite objective loss
- Porphyria (acute intermittent): episodic attacks of neuropathy with abdominal pain, psychiatric symptoms, urine that turns red/dark on standing
- Fabry's disease: X-linked, burning/lancinating pain in hands and feet in males since childhood, angiokeratomas around umbilicus and scrotum, premature cardiovascular disease
- Familial amyloid polyneuropathy: autonomic + sensory neuropathy, family history, carpal tunnel syndrome, cardiomyopathy
Rules out acquired causes when hereditary pattern is clear, preventing unnecessary workup.
Question 7: Are There Associated Medical Conditions?
This is the cornerstone of etiologic diagnosis. Systematically ask about:
Metabolic/endocrine:
- Diabetes mellitus (most common cause, ~50% of polyneuropathies)
- Prediabetes/metabolic syndrome (accounts for many "idiopathic" neuropathies)
- Hypothyroidism, uraemia, liver disease, vitamin deficiencies (B1, B6, B12, folate, vitamin E)
Malignancy:
- Known cancer (especially lung, breast, lymphoma) → paraneoplastic neuropathy (anti-Hu antibodies); may precede tumour diagnosis
- Multiple myeloma, POEMS syndrome → paraproteinemic neuropathy
Autoimmune/connective tissue:
- Rheumatoid arthritis, lupus, Sjögren's, vasculitis, sarcoidosis
- Sjögren's → sensory neuronopathy (ganglionopathy) or painful sensory neuropathy
Infections:
- HIV (distal painful sensory neuropathy, most common neurological complication)
- Leprosy (most common treatable neuropathy worldwide; thickened cutaneous nerves)
- Lyme disease (mononeuropathy multiplex, cranial nerve palsies)
- Hepatitis B/C (cryoglobulinemia → vasculitic mononeuropathy multiplex)
- CMV, EBV (GBS trigger, polyradiculopathy in immunocompromised)
Toxic/drug history (critical - ask about every medication and exposure):
- Drugs: chemotherapy (vincristine, cisplatin, paclitaxel), isoniazid, metronidazole, nitrofurantoin, phenytoin, thalidomide, amiodarone, colchicine
- Alcohol (direct toxic + nutritional B1 deficiency)
- Heavy metals: lead (motor-predominant), arsenic (painful sensory), thallium (alopecia + neuropathy)
- Industrial solvents: n-hexane, methyl n-butyl ketone
Rules out:
- If a clear toxic/drug cause identified → acquired toxic neuropathy (remove agent, observe for recovery)
- If DM present and neuropathy is distal, symmetrical, sensory-predominant → likely diabetic polyneuropathy, but UMN signs or rapid course mandate exclusion of other causes
C. Additional History Points
Social history: alcohol use (amount and duration), occupation (solvent/heavy metal exposure), dietary habits (veganism → B12 deficiency)
Preceding illness: URI or diarrhoea 2-4 weeks before weakness onset → GBS (Campylobacter jejuni most common trigger)
Review of systems: carpal tunnel (common in amyloid, hypothyroidism, acromegaly, diabetes), hearing loss (CMT-X, Refsum's disease), optic atrophy (CMT2 variants), skin changes (leprosy, Fabry's angiokeratomas), orthostatic symptoms
PART II: CLINICAL EXAMINATION
A. General Inspection
Before touching the patient:
- Pes cavus (high arched foot) + hammer toes → hereditary neuropathy (CMT)
- Foot drop gait (steppage gait) → distal motor neuropathy
- Wide-based ataxic gait → severe proprioceptive loss (sensory ataxia) - do the patient's symptoms worsen in the dark? This distinguishes sensory from cerebellar ataxia
- Fasciculations in muscle → suggests motor neuron disease rather than pure peripheral neuropathy
- Skin: leprosy patches (hypopigmented, anaesthetic), angiokeratomas (Fabry's), xanthomas (hyperlipidaemia)
- Thickened nerves: leprosy, CMT, amyloid, Refsum's disease - palpate greater auricular, ulnar at elbow, common peroneal at fibular head
B. Motor Examination
Inspection:
- Wasting: distal > proximal distribution in polyneuropathy; "inverted champagne bottle" legs (below-knee atrophy) in CMT
- Fasciculations: ALS > peripheral neuropathy
Power testing (MRC grading 0-5):
- Test distal muscles first: ankle dorsiflexors (tibialis anterior), intrinsic foot/hand muscles, extensor hallucis longus
- Then proximal: hip flexors, shoulder abductors
- Predominantly distal weakness = axonal polyneuropathy
- Proximal + distal weakness = demyelinating polyneuropathy (GBS, CIDP) - this is a key differential finding
- Asymmetric or multifocal weakness = mononeuropathy multiplex (vasculitis), radiculopathy, plexopathy
Rules out:
- Proximal-predominant weakness without sensory loss → myopathy (check CK, EMG)
- Global weakness with fatigability (worse with repetition, worse in afternoon) → NMJ disorder (myasthenia gravis)
- UMN signs (spasticity, clonus, extensor plantar) with weakness → central/combined lesion, not pure PNS
C. Sensory Examination
Test each modality separately, as selective loss distinguishes fiber types:
Large-fiber modalities (A-beta fibres):
- Vibration (128 Hz tuning fork): start at great toe; if absent, move to medial malleolus, tibial tuberosity, anterior iliac spine (to define the level)
- Joint position sense (proprioception): small passive movements of great toe; if impaired, confirm pseudoathetosis with eyes closed
- Light touch (cotton wool or 10g monofilament)
- Two-point discrimination
Small-fiber modalities (C and A-delta fibres):
- Pin-prick (sterile pin or neurological pin): compare left vs right, proximal vs distal
- Temperature (warm/cold test tubes or Tip-Therm)
Interpretation:
| Sensory loss pattern | Likely lesion |
|---|---|
| Loss of pain + temperature, preserved vibration/proprioception | Small-fiber neuropathy (DM, amyloid, HIV, idiopathic) |
| Loss of vibration + proprioception, preserved pain/temperature | Large-fiber neuropathy (B12 deficiency, CIDP, CMT) |
| All modalities lost distally ("glove and stocking") | Length-dependent polyneuropathy (most common pattern) |
| Severe proprioceptive loss with preserved strength | Sensory neuronopathy/ganglionopathy (paraneoplastic, Sjögren's, cisplatin) |
| Loss in dermatomal (band-like) distribution | Radiculopathy, NOT polyneuropathy |
| Loss along peripheral nerve territory | Mononeuropathy (e.g., carpal tunnel, ulnar nerve compression) |
| Dissociated sensory loss at a specific spinal level | Spinal cord lesion (syringomyelia, transverse myelitis) - rules out peripheral neuropathy |
Romberg test: patient stands with feet together, eyes open then closed
- Falls/sway only with eyes closed = sensory ataxia (posterior column or large-fiber neuropathy)
- Falls with eyes open also = cerebellar ataxia or vestibular disorder
D. Reflexes
Deep tendon reflexes are perhaps the single most important sign:
- Absent or reduced ankle jerks = peripheral neuropathy (almost universal finding); absent throughout = more widespread neuropathy
- Loss in an asymmetric or patchy pattern = mononeuropathy multiplex, radiculopathy
- Preserved or brisk reflexes in the presence of weakness or sensory loss = question whether lesion is truly peripheral; if UMN signs coexist → combined system disease (B12), ALS, or structural cord lesion
Rules out:
- Hyperreflexia/clonus/Babinski = UMN lesion (cord or brain), NOT peripheral neuropathy
- Selective loss of knee jerks with preserved ankle jerks = L3/L4 radiculopathy rather than peripheral neuropathy
Plantar responses: should be flexor (downgoing) in peripheral neuropathy; extensor response (Babinski) = UMN sign, pointing to spinal cord or brain
E. Autonomic Examination
- Blood pressure lying and standing: orthostatic hypotension (fall ≥20 mmHg systolic) without reflex tachycardia = autonomic neuropathy (DM, amyloid, GBS, HSAN)
- Skin: anhidrosis, trophic changes in feet, vasomotor changes (cold/cyanotic feet)
- Pupils: Argyll Robertson pupils or Horner's syndrome in autonomic neuropathy
- Bladder and bowel: palpate for distended bladder; rectal tone
Rules out:
- Orthostatic hypotension with appropriate tachycardia = hypovolaemia or autonomic failure from a non-neurological cause
- Absent sweating over trunk with preserved limb sweating = central autonomic lesion, not peripheral
F. Cranial Nerve Examination
Cranial nerve involvement helps narrow the diagnosis:
- Facial diplegia (bilateral VII) = GBS (occurs in >50% of cases), Lyme disease, sarcoidosis
- Ophthalmoplegia (III, IV, VI) + ataxia + areflexia = Miller Fisher syndrome (GBS variant, anti-GQ1b antibodies)
- Bilateral lower cranial nerve palsies (IX, X, XII: dysphagia, dysarthria) = GBS, ALS, myasthenia gravis
- Trigeminal sensory neuropathy = Sjögren's syndrome, paraneoplastic
- Hearing loss with neuropathy = Refsum's disease (also has retinitis pigmentosa + ataxia), CMT-X
G. Special Tests on Examination
- Tinel's sign (wrist, elbow): focal entrapment neuropathies (carpal tunnel, cubital tunnel)
- Phalen's test: carpal tunnel syndrome
- Straight leg raise: lumbosacral radiculopathy (rules out polyneuropathy at that level)
- Nerve palpation: thickened, palpable nerves at ulnar at elbow, common peroneal at fibular head = CMT, leprosy, amyloid
- Tandem gait, heel-toe walking: discloses subtle proximal weakness or ataxia
- Pseudoathetosis: slow writhing movements of outstretched fingers with eyes closed = severe proprioceptive loss
Summary: Key Differentiating Features at a Glance
| Feature | Peripheral Neuropathy | UMN/Cord Lesion | Myopathy | NMJ Disorder |
|---|---|---|---|---|
| Reflexes | Reduced/absent | Increased (Babinski+) | Normal/reduced | Normal |
| Plantar response | Flexor | Extensor | Flexor | Flexor |
| Weakness distribution | Distal > proximal (or diffuse) | Pyramidal pattern | Proximal > distal | Bulbar, ocular, proximal |
| Sensation | Impaired (glove/stocking) | Dermatomal level | Normal | Normal |
| Wasting | Present (late) | Absent early | Present | Absent |
| Fatigability | No | No | Rare | Yes (myasthenia) |
| Autonomic signs | Possible | Possible (cord) | No | No |
| Gait | Steppage or ataxic | Spastic-scissor | Trendelenburg/waddling | Variable |
Topographic Patterns and Their Differential Diagnosis (Adams & Victor + Harrison's)
| Pattern | Key Clinical Features | Causes to Consider |
|---|---|---|
| Symmetric distal polyneuropathy | Stocking-glove sensory loss, distal areflexia, minimal weakness | DM, idiopathic (CSPN), toxic, alcohol, B12 deficiency, CMT, amyloid |
| Acute symmetric ascending motor + sensory | Rapid ascent, areflexia, facial diplegia, autonomic instability | GBS |
| Chronic symmetric proximal + distal | Proximal AND distal weakness, sensory loss | CIDP, paraproteinaemia, lymphoma |
| Asymmetric multifocal (mononeuropathy multiplex) | Multiple individual nerve territories, stepwise progression | Vasculitis, diabetes, leprosy, sarcoid, cryoglobulinaemia, Lyme |
| Sensory ataxia, severe proprioceptive loss | Romberg+, pseudoathetosis, intact strength | Paraneoplastic (anti-Hu), Sjögren's, cisplatin, CANVAS, B6 toxicity |
| Pure motor neuropathy | No sensory symptoms/signs | MMN (with conduction block), GBS, porphyria |
| Small-fiber neuropathy | Burning pain, allodynia, normal NCVs, preserved reflexes | DM/prediabetes, amyloid, idiopathic, HIV |
References
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 457: "Peripheral Neuropathy," pp. 3638-3645
- Adams and Victor's Principles of Neurology, 12th Ed., Chapter 43: "Approach to the Patient with Peripheral Neuropathy," pp. 1295-1298
- Goldman-Cecil Medicine, Chapter: "Peripheral Neuropathy," pp. 4073+
This is a shared conversation. Sign in to Orris to start your own chat.