Prepare a pathology-based summary of the topic Von Willebrand disease, explaining the cause, pathogenesis, clinical features, lab test findings, any peripheral smear findings, the types, sources of von Willebrand factor, a short summary of treatment required for NEET-PG 2026. This should help me answer almost 100% of questions that come from the pathology aspect of von Willebrand disease. It should also give the reason for a particular statement such that I don't have to mug it up, I can understand it, but it should be short and precise.
Von Willebrand Disease (VWD) — Pathology Summary for NEET-PG 2026
1. Basics & Inheritance
- Most common inherited bleeding disorder (prevalence ~1% of the general population; clinically significant bleeding ~1:10,000)
- Autosomal dominant (most types); Type 3 is autosomal recessive
- Affects both sexes equally (autosomal) — key distinguisher from hemophilia (X-linked)
- Gene: chromosome 12 (vWF gene)
2. Sources of vWF
| Source | Storage Granule | Released by |
|---|---|---|
| Vascular endothelial cells (primary source) | Weibel-Palade bodies | Thrombin, DDAVP, shear stress |
| Megakaryocytes / Platelets | α-granules | Platelet activation |
Why it matters: DDAVP works by triggering endothelial release from Weibel-Palade bodies — this is why it helps in Type 1 (mild) VWD but NOT in Type 3 (no vWF to release).
3. Normal Functions of vWF
- Platelet adhesion: vWF binds exposed subendothelial collagen at sites of injury, then acts as a bridge to platelet GpIb receptor → platelet adhesion (primary hemostasis)
- Carrier/chaperone for Factor VIII: vWF binds and protects Factor VIII from premature degradation in plasma; when vWF is deficient, Factor VIII gets rapidly cleared → reduced Factor VIII levels
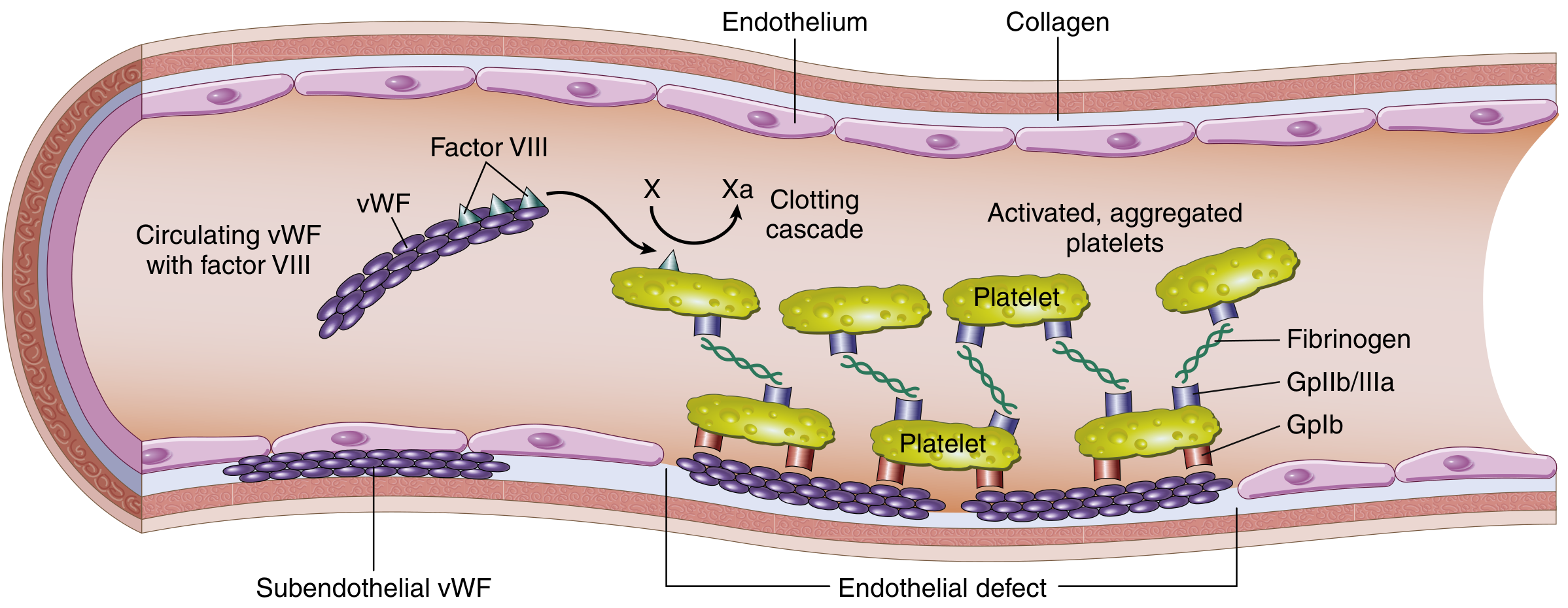
Why vWF deficiency causes prolonged bleeding time: Loss of platelet adhesion (GpIb–vWF–collagen axis). This is a primary hemostasis defect, hence mucosal bleeding predominates.
4. Pathogenesis
- Reduced quantity of vWF (quantitative defect — Types 1 & 3), or
- Structurally/functionally abnormal vWF (qualitative defect — Type 2)
5. Clinical Features
| Feature | Reason |
|---|---|
| Mucocutaneous bleeding (epistaxis, gum bleeding, GI bleed, heavy menses) | Primary hemostasis defect — platelet plug formation fails |
| Easy bruising | Same mechanism |
| Prolonged bleeding after cuts/surgery | Failure of platelet adhesion + mildly reduced FVIII |
| Menorrhagia | Very common presenting complaint in females |
| Hemarthrosis / deep muscle hematoma | Rare; only in Type 3 (severe FVIII deficiency — mimics hemophilia A) |
The bleeding pattern is mucocutaneous (like thrombocytopenia/platelet disorders) — NOT deep tissue/hemarthrosis (which is coagulation disorder pattern) — except in Type 3 where FVIII is severely reduced.
6. Types of VWD
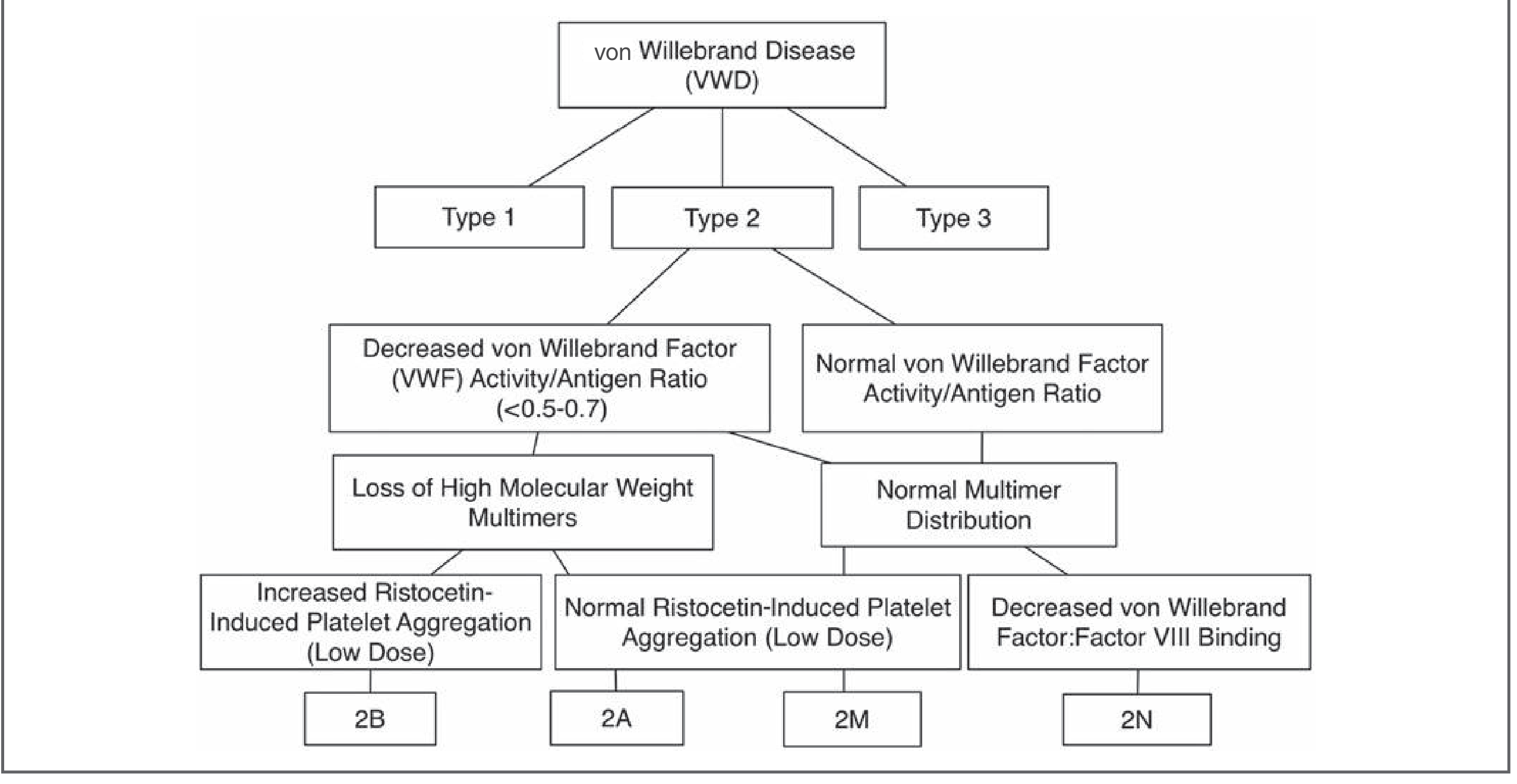
Type 1 — Partial Quantitative Deficiency (Most common — 75% of cases)
- Autosomal dominant
- Reduced quantity of vWF, but structurally normal; entire multimer spectrum present but in decreased amounts
- Mildly decreased Factor VIII
- Mild bleeding symptoms
- Responds well to DDAVP
Type 2 — Qualitative Defects (Abnormal vWF)
| Subtype | Defect | HMWMs | RIPA (low dose ristocetin) | Key Feature |
|---|---|---|---|---|
| 2A | Decreased synthesis OR increased proteolysis of HMWMs | Absent | Decreased/Absent | Decreased function; decreased ratio vWF:RCo/vWF:Ag <0.6 |
| 2B | Gain-of-function mutation → vWF binds GpIb too avidly | Absent (consumed) | Increased (aggregation at low dose ~0.3–0.5 mg/mL) | Mild thrombocytopenia (platelets consumed by spontaneous aggregation); most dangerous subtype — DDAVP contraindicated |
| 2M | Loss-of-function mutation in platelet-binding site | Present (normal) | Decreased | Normal multimer pattern but decreased function |
| 2N (Normandy) | Mutation in vWF's FVIII-binding site | Normal | Normal | Markedly low Factor VIII despite normal vWF antigen — mimics hemophilia A; autosomal inheritance distinguishes it |
Memory hook for 2B: "B = Binds Better" → vWF is hyperactive → consumes HMWMs and platelets → thrombocytopenia. Memory hook for 2N: "N = No FVIII binding" → FVIII is unprotected → rapidly destroyed → looks like Hemophilia A, but autosomal.
Type 3 — Severe/Complete Quantitative Deficiency
- Autosomal recessive (homozygous)
- Near-complete absence of vWF
- Severely low Factor VIII (because no carrier protein)
- Clinically resembles hemophilia A — hemarthrosis, deep hematomas
- Does NOT respond to DDAVP (no vWF to release)
- Requires Factor VIII concentrate or vWF-containing concentrates
7. Laboratory Findings
Screening Tests
| Test | Result in VWD | Reason |
|---|---|---|
| Bleeding Time (BT) | Prolonged | Platelet adhesion defect |
| Platelet count | Usually normal (↓ in Type 2B) | Not thrombocytopenic (except 2B) |
| PT | Normal | Extrinsic pathway unaffected |
| aPTT | Normal or mildly prolonged | FVIII is mildly reduced; prolonged notably in Type 3 and 2N |
| Platelet function analyzer (PFA-100) | Prolonged closure time | Sensitive test for platelet adhesion defects |
Classic VWD lab pattern: Normal platelet count + Prolonged BT + Normal PT + Normal or slightly prolonged aPTT. If aPTT is markedly prolonged → Type 3 or Type 2N.
Specific/Confirmatory Tests
| Test | What it measures | Findings |
|---|---|---|
| vWF antigen (vWF:Ag) | Total vWF protein (quantity) | Decreased in Types 1, 3; Normal in 2N, 2M |
| Ristocetin cofactor activity (vWF:RCo) | vWF function — ability to bind platelet GpIb | Decreased in most types |
| vWF:RCo / vWF:Ag ratio | Functional adequacy of vWF | <0.6 in Type 2A, 2B; normal in Type 1 and 3 |
| Factor VIII activity (FVIII:C) | FVIII level | Mildly ↓ in Types 1 & 2; markedly ↓ in Type 3 & 2N |
| Ristocetin-induced platelet aggregation (RIPA) | Platelet aggregation with ristocetin | ↑ at low dose in Type 2B; ↓/absent in 2A and 2M |
| vWF multimer analysis (SDS-agarose gel electrophoresis) | Size distribution of vWF multimers | Gold standard for subtyping Type 2; absence of HMWMs in 2A and 2B |
| vWF:FVIII binding assay | vWF's ability to bind Factor VIII | Markedly decreased in Type 2N |
Ristocetin is an antibiotic that mimics exposed subendothelium — it forces vWF to bind platelet GpIb. If vWF is absent or dysfunctional, platelets fail to aggregate. In Type 2B, even tiny amounts of ristocetin trigger aggregation because the vWF is hyperactive.
Peripheral Blood Smear
- Usually normal
- Platelet morphology is normal (VWD is a plasma protein defect, not a platelet structural defect)
- Mild thrombocytopenia may be seen in Type 2B (from platelet consumption), but platelets appear morphologically normal
- No schistocytes, no giant platelets (distinguishes from BSS/TTP)
8. Differentiating VWD from Hemophilia A
| Feature | VWD (Type 1/2) | Hemophilia A |
|---|---|---|
| Inheritance | Autosomal dominant | X-linked recessive |
| Sex | Both sexes | Males primarily |
| Bleeding type | Mucocutaneous | Deep (hemarthrosis, muscle) |
| BT | Prolonged | Normal |
| PT | Normal | Normal |
| aPTT | Normal or mildly prolonged | Prolonged |
| FVIII | Mildly reduced | Markedly reduced |
| vWF:Ag | Reduced | Normal |
| RIPA | Reduced | Normal |
Type 2N VWD exception: Presents like Hemophilia A (↑↑ aPTT, markedly low FVIII) but BT is prolonged and inheritance is autosomal — these two features point away from hemophilia.
9. Treatment
| Situation | Treatment | Mechanism |
|---|---|---|
| Type 1 (mild-moderate), pre-procedure | DDAVP (desmopressin) IV/intranasal | Releases vWF from endothelial Weibel-Palade bodies → acute rise in vWF and FVIII |
| Type 3, severe Type 1, major surgery | vWF-containing Factor VIII concentrates (e.g., Humate-P, Alphanate) | Replaces deficient vWF directly |
| Type 2B | DDAVP is CONTRAINDICATED | Would release more abnormal hyperactive vWF → worsens thrombocytopenia via platelet consumption |
| Adjunct | Tranexamic acid / ε-aminocaproic acid | Antifibrinolytics — stabilize clot; useful for mucosal bleeding |
| Menorrhagia | Oral contraceptives + tranexamic acid | Estrogens increase vWF levels |
| Type 2N / hemophilia-like | Factor VIII concentrate + vWF | Replaces both deficient factors |
Why DDAVP works: It is a vasopressin analogue that acts on V2 receptors on endothelial cells, triggering exocytosis of Weibel-Palade bodies → releases stored vWF and tPA. Why cryoprecipitate is used: Contains high concentration of vWF, FVIII, fibrinogen — older option, still used when concentrates unavailable.
10. High-Yield One-Liners for NEET-PG
- Most common inherited bleeding disorder = VWD (not hemophilia)
- vWF stored in: Weibel-Palade bodies (endothelium) + α-granules (platelets)
- vWF links platelets to collagen via: GpIb receptor
- Classic lab: Normal platelet count + Prolonged BT + Normal PT + Normal/slightly prolonged aPTT
- Type 2B = gain-of-function + HMWMs absent + thrombocytopenia + increased RIPA at low dose ristocetin
- Type 2N = mimics Hemophilia A + autosomal + normal vWF:Ag + normal RIPA + markedly low FVIII
- DDAVP contraindicated in Type 2B (worsens thrombocytopenia)
- Type 3 = most severe + autosomal recessive + mimics Hemophilia A + requires FVIII/vWF concentrate
- Peripheral smear = normal (no schistocytes, no giant platelets)
- Blood group O individuals have ~25% lower vWF levels — can simulate Type 1 VWD
- Acquired VWD — causes: aortic stenosis (shear stress destroys HMWMs), lymphoproliferative disorders (antibodies), hypothyroidism (decreased synthesis)
- Ristocetin → tests vWF–GpIb axis; absent aggregation in VWD; hyperaggregation at LOW dose in Type 2B
Convert the above info on von Willebrand disease to a very high-yield clinical-based Anki cards to be imported to AnkiDroid app. It should cover almost 100% of pathology questions that can be asked for NEET PG 2026 exam. It should help to answer real clinical questions that can be asked for the exam. It should be in text format so that I can copy and paste to a text app and then import to AnkiDroid app. In the end, it should also give me a summary in the form of one single Anki card in the same format with proper spacing and punctuation.
TAB Back), ready to copy-paste into a .txt file and import into AnkiDroid (Basic note type, fields separated by tab).Von Willebrand Disease — NEET PG 2026 | Anki Deck
#separator:tab
#html:false
#notetype:Basic
#deck:Pathology::Von Willebrand Disease
What is the most common inherited bleeding disorder? Von Willebrand Disease (VWD). Prevalence ~1% of population; clinically significant bleeding ~1:10,000.
What is the inheritance pattern of Von Willebrand Disease? Autosomal Dominant (most types). Type 3 is Autosomal Recessive. Both sexes affected equally — key difference from Hemophilia A (X-linked recessive, males only).
Where is vWF synthesized and stored? 1. Vascular endothelial cells → stored in Weibel-Palade bodies (primary source). 2. Megakaryocytes/Platelets → stored in alpha-granules. Released by: thrombin, DDAVP, shear stress.
What are the two main functions of vWF? 1. Platelet adhesion: vWF bridges exposed subendothelial collagen to platelet GpIb receptor → forms primary platelet plug. 2. Carrier/Chaperone for Factor VIII: protects FVIII from degradation in plasma. Deficiency of vWF → secondary FVIII reduction.
Why does vWF deficiency cause prolonged bleeding time? vWF is required for platelet adhesion to injured subendothelium (via GpIb receptor). Its absence = primary hemostasis defect = mucocutaneous bleeding + prolonged bleeding time. PT remains normal (extrinsic pathway unaffected).
What is the platelet receptor that binds vWF? GpIb (Glycoprotein Ib). vWF acts as bridge: Collagen → vWF → GpIb → Platelet adhesion. (Aggregation uses GpIIb/IIIa + Fibrinogen — separate step.)
A patient has epistaxis, menorrhagia, and easy bruising since childhood. Both mother and sister have similar complaints. Most likely diagnosis? Von Willebrand Disease Type 1. Autosomal dominant → family history in both sexes. Mucocutaneous bleeding pattern = primary hemostasis defect (vWF-related).
What type of bleeding does VWD typically cause and why? Mucocutaneous bleeding: epistaxis, gum bleeding, menorrhagia, GI bleed, easy bruising. Reason: platelet adhesion defect (vWF–GpIb axis failure) = primary hemostasis disorder. Deep tissue/hemarthrosis only in Type 3 (severe FVIII deficiency).
What is the classic lab pattern in VWD? Normal platelet count + Prolonged Bleeding Time + Normal PT + Normal or mildly prolonged aPTT. PFA-100 closure time is prolonged (sensitive test for platelet adhesion defects).
Why is the PT normal in VWD? PT tests the extrinsic pathway (Factor VII, X, V, II, fibrinogen). vWF is not part of this pathway. PT is unaffected in VWD.
Why is aPTT prolonged (or normal) in VWD? aPTT tests intrinsic pathway including Factor VIII. vWF carries and protects FVIII. In VWD: mild vWF reduction → mild FVIII reduction → aPTT slightly prolonged or normal. In Type 3 or 2N: FVIII markedly reduced → aPTT markedly prolonged.
What is the peripheral blood smear finding in VWD? Usually NORMAL. Platelet count normal, platelet morphology normal. Mild thrombocytopenia only in Type 2B (due to platelet consumption by hyperactive vWF). No schistocytes (no microangiopathy).
What are the initial lab tests for VWD diagnosis? Three initial tests: 1. vWF antigen (vWF:Ag) — quantity. 2. vWF Ristocetin Cofactor activity (vWF:RCo) — function. 3. Factor VIII activity (FVIII:C) — indirectly measures vWF chaperone function.
What does ristocetin do and why is it used in VWD testing? Ristocetin is an antibiotic that mimics exposed subendothelium → forces vWF to bind platelet GpIb → causes platelet aggregation. In VWD: vWF absent or dysfunctional → no/reduced aggregation (RIPA decreased). Tests the vWF–GpIb axis.
What is the vWF:RCo/vWF:Ag ratio and what does <0.6 indicate? This ratio compares vWF function to its quantity. Normal ≥0.6. Ratio <0.6 = qualitative defect → vWF present but functionally abnormal → Type 2A or 2B. In Type 1: ratio normal (both quantity and function equally reduced). In Type 3: both very low (ratio may be roughly normal or unreliable).
What is the gold standard test for subtyping VWD Type 2? vWF Multimer Analysis by SDS-agarose gel electrophoresis + Western blot. Shows size distribution of vWF multimers. Types 2A and 2B: loss of high-molecular-weight multimers (HMWMs). Type 2M: normal multimer pattern despite decreased function. Type 2N: normal multimer pattern.
What is the three-step supplementary testing for Type 2 VWD classification? 1. vWF Multimer Analysis — confirms loss of HMWMs (Types 2A, 2B, 2M). 2. RIPA at low-dose ristocetin (~0.5 mg/mL) — positive (increased aggregation) in Type 2B. 3. vWF:FVIII binding assay — decreased in Type 2N.
What is Type 1 VWD? Most common type (75% of cases). Autosomal dominant. Partial QUANTITATIVE deficiency — vWF reduced in quantity but qualitatively normal. Full multimer spectrum present but in decreased amounts. Mild bleeding. vWF:RCo/vWF:Ag ratio ≥0.6. Responds well to DDAVP.
What is Type 2A VWD? Qualitative defect. Loss of high-molecular-weight multimers (HMWMs) due to: (a) impaired multimerization, or (b) increased susceptibility to ADAMTS13 proteolysis. Decreased vWF:RCo and decreased RIPA. Ratio vWF:RCo/vWF:Ag <0.6. HMWMs absent on multimer analysis. Treat with vWF concentrates (not DDAVP alone).
What is Type 2B VWD and what is its unique complication? Gain-of-function mutation → vWF binds platelet GpIb TOO avidly → spontaneous consumption of HMWMs and platelets. Key features: absent HMWMs + MILD THROMBOCYTOPENIA + increased RIPA at LOW-DOSE ristocetin (~0.3–0.5 mg/mL). Ratio <0.6. DDAVP is CONTRAINDICATED (releases more abnormal vWF → worsens thrombocytopenia).
Why is DDAVP contraindicated in Type 2B VWD? DDAVP releases stored vWF from Weibel-Palade bodies. In Type 2B, the vWF released is hyperactive (gain-of-function) → binds platelets even more → worsens platelet consumption → worsens thrombocytopenia.
What is Type 2M VWD? Qualitative defect with NORMAL multimer distribution (HMWMs present). Loss-of-function mutation in vWF's platelet-binding site → decreased vWF:RCo/vWF:Ag ratio (<0.6). RIPA at low dose: normal (no hyperactivation). Resembles Type 2A clinically but multimers are normal — key distinguishing feature.
What is Type 2N (Normandy) VWD? Mutation in vWF's FVIII-binding site → vWF cannot carry/protect FVIII → FVIII rapidly degraded. Lab: normal vWF:Ag + normal RIPA + markedly LOW FVIII:C. Mimics Hemophilia A. Key distinguisher: AUTOSOMAL inheritance (not X-linked); bleeding time is prolonged; vWF:FVIII binding assay confirms (decreased binding).
How do you differentiate Type 2N VWD from Hemophilia A? Type 2N VWD: Autosomal inheritance (both sexes affected) + prolonged bleeding time + normal vWF:Ag + markedly low FVIII. Hemophilia A: X-linked recessive (males only) + normal bleeding time + normal vWF:Ag + markedly low FVIII. Confirm 2N with vWF:FVIII binding assay (decreased).
What is Type 3 VWD? Most severe type. Autosomal RECESSIVE (homozygous). Near-COMPLETE absence of vWF → severely low FVIII (no carrier). Presents like Hemophilia A: hemarthrosis, deep hematomas + mucocutaneous bleeding. Markedly prolonged aPTT + prolonged BT. DDAVP ineffective (no vWF to release). Treat with vWF-containing FVIII concentrates.
Which type of VWD has thrombocytopenia? Type 2B. Gain-of-function vWF binds platelets spontaneously → platelet consumption → mild to moderate thrombocytopenia. All other types have NORMAL platelet count.
A patient presents with mild thrombocytopenia, absence of high-molecular-weight multimers, and platelet aggregation at LOW-dose ristocetin. What is the diagnosis? Type 2B Von Willebrand Disease. Gain-of-function mutation → hyperactive vWF → spontaneous platelet consumption → thrombocytopenia + loss of HMWMs + RIPA positive at low dose. DDAVP contraindicated.
A male patient has autosomal family history of 'hemophilia-like' bleeding. Lab shows markedly low FVIII but normal vWF:Ag and normal RIPA. Diagnosis? Type 2N (Normandy) VWD. Mutation in vWF's FVIII-binding domain → unprotected FVIII degraded → low FVIII. Normal vWF:Ag. Autosomal inheritance (rules out true Hemophilia A). Confirm with vWF:FVIII binding assay.
A woman has severe bleeding since childhood, hemarthrosis, prolonged aPTT, prolonged BT, nearly absent vWF. Diagnosis and treatment? Type 3 VWD. Autosomal recessive. Complete vWF deficiency → severe FVIII deficiency → hemophilia-like picture. Treatment: vWF-containing Factor VIII concentrates (e.g., Humate-P). DDAVP is ineffective.
What is the treatment for Type 1 VWD (mild bleeding/pre-procedure)? DDAVP (Desmopressin) — IV or intranasal. Mechanism: acts on V2 receptors on endothelial cells → exocytosis of Weibel-Palade bodies → acute release of stored vWF and tPA → raises vWF and FVIII levels within 30–60 min.
What is the treatment for severe VWD or Type 3? vWF-containing Factor VIII concentrates: e.g., Humate-P, Alphanate. These provide both vWF and FVIII. Cryoprecipitate (older option) — contains vWF, FVIII, fibrinogen, fibronectin.
What adjunct treatments are used for mucosal bleeding in VWD? Antifibrinolytics: Tranexamic acid or Epsilon-aminocaproic acid. Stabilize formed clot by inhibiting fibrinolysis. Useful for epistaxis, menorrhagia, dental procedures. Oral contraceptives in women with menorrhagia (estrogens increase vWF levels).
Why do estrogens help in VWD? Estrogens increase synthesis and secretion of vWF from endothelial cells. Hence oral contraceptive pills (OCPs) reduce menorrhagia in VWD patients. Also explains why VWD symptoms may improve during pregnancy (estrogen/progesterone rise).
What is the effect of blood group O on vWF levels? Blood group O individuals have ~25% LOWER vWF levels than non-O blood groups (due to increased vWF glycosylation and clearance). This can mimic Type 1 VWD — always check ABO group when interpreting borderline vWF:Ag levels.
What is Acquired Von Willebrand Syndrome (AVWS)? VWD-like syndrome developing in adults with NO personal or family history of lifelong bleeding. Causes: 1. Autoantibodies (lymphoproliferative disorders, myeloma, MGUS). 2. Adsorption onto cells (Wilms tumor with GPIb expression, myeloproliferative disorders with high platelet count). 3. Increased proteolysis by shear stress (aortic stenosis, ventricular assist devices, congenital heart disease). 4. Hypothyroidism (decreased vWF synthesis).
Aortic stenosis causes what type of hemostatic defect? Acquired Von Willebrand Syndrome — specifically loss of high-molecular-weight vWF multimers due to high shear stress across the stenotic valve → ADAMTS13-mediated proteolysis of HMWMs. This is why aortic stenosis is associated with GI angiodysplasia bleeding (Heyde's syndrome).
How does VWD differ from Glanzmann Thrombasthenia in lab findings? Both: prolonged BT + normal PT + normal aPTT + normal platelet count. Difference: VWD — decreased vWF:Ag, decreased RIPA (ristocetin fails). Glanzmann — normal vWF, RIPA normal (vWF–GpIb axis intact); defective GpIIb/IIIa → ADP/collagen/thrombin aggregation absent. Clot retraction absent in Glanzmann; normal in VWD.
How does VWD differ from Bernard-Soulier Syndrome? Both: prolonged BT. Bernard-Soulier: deficiency of GpIb receptor → RIPA absent (like VWD). BUT: Bernard-Soulier has THROMBOCYTOPENIA + GIANT PLATELETS (macrothrombocytopenia) on smear — absent in VWD. vWF:Ag is NORMAL in Bernard-Soulier.
What is RIPA and how does it differ between Type 2A, 2B, and 2M? RIPA = Ristocetin-Induced Platelet Aggregation. 2A: DECREASED at both normal and low dose (HMWMs absent, low function). 2B: INCREASED at LOW dose (~0.3–0.5 mg/mL) — hyperactive vWF aggregates platelets even with tiny ristocetin. 2M: DECREASED (like 2A) but multimers are NORMAL. Key: only 2B gives increased aggregation at low dose.
Which types of VWD are QUANTITATIVE and which are QUALITATIVE? Quantitative (amount reduced): Types 1 and 3. Qualitative (structurally/functionally abnormal): Types 2A, 2B, 2M, 2N.
What is the multimer pattern in each VWD type? Type 1: All multimers present, decreased in amount. Type 2A: Loss of HMWMs (absent). Type 2B: Loss of HMWMs (consumed by platelets). Type 2M: NORMAL multimer distribution (function reduced, not size). Type 2N: NORMAL multimer distribution (FVIII binding impaired, not multimer structure). Type 3: Near-complete absence of all multimers.
What does DDAVP stand for and how does it work in VWD? DDAVP = Desmopressin (1-deamino-8-D-arginine vasopressin). Acts on V2 receptors on vascular endothelial cells → exocytosis of Weibel-Palade bodies → releases stored vWF (and tPA) into circulation → raises vWF and FVIII levels acutely. Used in Type 1 (mainstay), cautiously in Type 2A/2M. Contraindicated in Type 2B.
What is cryoprecipitate and when is it used in VWD? Cryoprecipitate = cold-insoluble fraction of fresh frozen plasma. Contains: vWF (high concentration), FVIII, fibrinogen, fibronectin, Factor XIII. Used when vWF-containing concentrates are unavailable. Older treatment option for severe VWD and Type 3.
A patient with VWD undergoes dental extraction. What prophylactic treatment is given? DDAVP (if Type 1) before the procedure + Tranexamic acid mouthwash/systemic (antifibrinolytic) post-procedure. For severe types: vWF-containing FVIII concentrate. Avoid aspirin and NSAIDs (impair platelet function further).
What is the confirmatory test for Type 2B VWD vs. Platelet-type (Pseudo) VWD? Both show: increased RIPA at low dose ristocetin + thrombocytopenia + loss of HMWMs. Differentiation: vWF:platelet binding assay (ELISA). In Type 2B: vWF is abnormal → decreased binding in ELISA. In Platelet-type VWD: GpIb on platelet is abnormal (not vWF) → normal vWF:platelet binding by ELISA.
What is the FVIII:C/vWF:Ag ratio significance in Type 2N? In Type 2N, FVIII is markedly low but vWF:Ag is normal → FVIII:C/vWF:Ag ratio is markedly DECREASED (<0.6). This pattern (low FVIII + normal vWF + autosomal inheritance) should trigger vWF:FVIII binding assay to confirm Type 2N VWD.
What is the most sensitive bedside/point-of-care test for VWD? Platelet Function Analyzer (PFA-100): measures platelet adhesion and aggregation under high shear stress using collagen/ADP and collagen/epinephrine cartridges. Closure time is prolonged in VWD. More sensitive than bleeding time and reproducible. Bleeding time is now largely replaced by PFA-100.
A 16-year-old female presents with menorrhagia since menarche. Her mother has similar history. Lab: normal platelet count, normal PT, slightly prolonged aPTT, prolonged PFA-100. Most likely diagnosis and next investigation? Von Willebrand Disease (Type 1, most likely). Next investigation: vWF:Ag + vWF:RCo + FVIII:C. Autosomal dominant pattern + mucocutaneous bleeding in a female = classic VWD presentation.
A patient with aortic stenosis develops recurrent GI bleeding. What is the mechanism? Heyde's Syndrome = acquired loss of high-molecular-weight vWF multimers due to high shear stress across stenotic aortic valve → ADAMTS13 cleaves HMWMs → acquired VWD type 2A-like pattern → impaired platelet adhesion → bleeding from GI angiodysplasia. Corrects after aortic valve replacement.
What is the gene locus for vWF? Chromosome 12p13. vWF gene is large (178 kb, 52 exons). There is also a non-functional pseudogene on chromosome 22. Mutations include missense, nonsense, deletions, and splice-site mutations.
Summary Card: Von Willebrand Disease — Complete High-Yield Overview DEFINITION: Most common inherited bleeding disorder; Autosomal dominant (Type 3 = AR). SOURCES OF vWF: Endothelium (Weibel-Palade bodies) + Platelets (alpha-granules). FUNCTIONS OF vWF: 1. Platelet adhesion via GpIb. 2. Carrier/protector of Factor VIII. CLINICAL FEATURES: Mucocutaneous bleeding (epistaxis, menorrhagia, easy bruising); Hemarthrosis only in Type 3. CLASSIC LAB: Normal platelet count + Prolonged BT/PFA-100 + Normal PT + Normal/mildly prolonged aPTT. TYPES: Type 1 = partial quantitative (most common, 75%; DDAVP works). Type 2A = no HMWMs, decreased RIPA, decreased ratio. Type 2B = hyperactive vWF, no HMWMs, thrombocytopenia, INCREASED low-dose RIPA, DDAVP CONTRAINDICATED. Type 2M = decreased function, NORMAL multimers. Type 2N = mimics Hemophilia A (low FVIII, normal vWF:Ag), autosomal. Type 3 = complete deficiency, AR, hemarthrosis, DDAVP ineffective. TREATMENT: DDAVP (Type 1 / mild) | vWF-containing FVIII concentrate (Type 3 / severe) | Tranexamic acid (adjunct) | DDAVP CONTRAINDICATED in Type 2B. KEY DISTINGUISHERS: VWD vs Hemophilia A: both sexes affected + prolonged BT in VWD. VWD vs Bernard-Soulier: giant platelets + thrombocytopenia in BSS. VWD vs Glanzmann: ADP/collagen aggregation absent in Glanzmann; RIPA absent in VWD.
How to import into AnkiDroid:
- Copy the entire text block above into a plain text file and save as
VWD_NEET_PG.txt - In AnkiDroid: tap ⋮ → Import (or use the desktop Anki to import, then sync)
- On desktop Anki: File → Import → select the .txt file
- Separator: Tab
- Notetype: Basic
- Deck:
Pathology::Von Willebrand Disease(or your preferred deck)
- The header lines (starting with
#) are automatically read as import settings by Anki and are not imported as cards
The last card ("Summary Card") is the single consolidated overview card you asked for — it contains every high-yield point in one card with proper field spacing and punctuation.
I only wanted 20 Anki cards, and the summary card should be a separate card.
#separator:tab
#html:false
#notetype:Basic
#deck:Pathology::Von Willebrand Disease
What is Von Willebrand Disease and what makes it unique among bleeding disorders? Most common inherited bleeding disorder (~1% prevalence). Autosomal dominant (Type 3 = AR). Affects BOTH sexes equally. Causes a COMPOUND defect: platelet adhesion failure (primary hemostasis) + secondary mild FVIII reduction (secondary hemostasis). Most symptoms come from the platelet defect.
Where is vWF synthesized and stored? 1. Vascular endothelial cells → Weibel-Palade bodies (primary source). 2. Megakaryocytes/Platelets → alpha-granules. Released by: DDAVP, thrombin, shear stress. (DDAVP works by triggering exocytosis of Weibel-Palade bodies.)
What are the two functions of vWF and what happens when it is deficient? 1. Platelet adhesion: bridges subendothelial collagen to platelet GpIb → primary hemostasis. 2. Carrier/protector of Factor VIII: unbound FVIII is rapidly degraded. Deficiency → mucocutaneous bleeding (platelet defect) + secondary FVIII reduction (coagulation defect, usually mild; severe only in Type 3).
What is the classic laboratory pattern in VWD? Normal platelet count + Prolonged bleeding time (PFA-100) + Normal PT + Normal or mildly prolonged aPTT. PT is normal because the extrinsic pathway is unaffected. aPTT is prolonged only when FVIII is significantly reduced (Type 3, Type 2N).
What are the three initial confirmatory tests for VWD? 1. vWF antigen (vWF:Ag) — measures quantity of vWF. 2. vWF Ristocetin Cofactor activity (vWF:RCo) — measures function (GpIb binding). 3. Factor VIII activity (FVIII:C) — reflects vWF chaperone function. vWF:RCo/vWF:Ag ratio <0.6 = qualitative (Type 2) defect.
What is ristocetin and why is it used in VWD testing? Ristocetin is an antibiotic that forces vWF to bind platelet GpIb → causes platelet aggregation in vitro (RIPA test). In VWD: vWF absent/dysfunctional → reduced or absent aggregation. Exception: Type 2B → increased aggregation even at LOW dose (~0.3–0.5 mg/mL) due to hyperactive vWF.
What is the peripheral blood smear finding in VWD? Usually NORMAL — normal platelet count and morphology (VWD is a plasma protein defect, not a platelet structural defect). Exception: Type 2B → mild thrombocytopenia (platelets consumed by hyperactive vWF), but platelet morphology is still normal. No schistocytes, no giant platelets.
What is Type 1 VWD? Most common (75% of cases). Autosomal dominant. Partial QUANTITATIVE deficiency — vWF reduced in amount but structurally normal. Full multimer spectrum present (just less of it). Ratio vWF:RCo/vWF:Ag is normal (≥0.6). Mild symptoms. Treatment: DDAVP (drug of choice).
What is Type 2A VWD? Qualitative defect. Loss of high-molecular-weight multimers (HMWMs) due to impaired synthesis OR increased ADAMTS13 proteolysis. Result: impaired platelet adhesion. RIPA decreased. Ratio vWF:RCo/vWF:Ag <0.6. HMWMs absent on multimer analysis. Treatment: vWF-containing concentrates (DDAVP less reliable).
What is Type 2B VWD and why is DDAVP contraindicated? Gain-of-function mutation → vWF binds GpIb too avidly → spontaneous platelet consumption → absent HMWMs + MILD THROMBOCYTOPENIA + increased RIPA at LOW-dose ristocetin. DDAVP releases more of the same hyperactive vWF → worsens platelet consumption → worsens thrombocytopenia. Treatment: vWF-containing concentrates.
What is Type 2M VWD and how does it differ from Type 2A? Both: decreased vWF function, decreased vWF:RCo/vWF:Ag ratio <0.6, decreased RIPA. Key difference: Type 2M has NORMAL multimer distribution (mutation affects platelet-binding site, not multimer structure). Type 2A has absent HMWMs. Multimer analysis is the distinguishing test.
What is Type 2N (Normandy) VWD and how does it mimic Hemophilia A? Mutation in vWF's FVIII-binding domain → FVIII unprotected → rapidly degraded → markedly low FVIII:C. Lab: normal vWF:Ag + normal RIPA + markedly low FVIII = identical to Hemophilia A. Key distinguishers from Hemophilia A: autosomal inheritance (both sexes affected) + prolonged bleeding time. Confirm with vWF:FVIII binding assay (decreased).
What is Type 3 VWD? Most severe. Autosomal RECESSIVE. Near-complete absence of vWF → severely low FVIII → clinically resembles Hemophilia A (hemarthrosis + deep hematomas) PLUS mucocutaneous bleeding. Markedly prolonged aPTT + prolonged BT. DDAVP ineffective (no vWF stores to release). Treatment: vWF-containing FVIII concentrates.
Summarize the multimer pattern across all VWD types. Type 1: All multimers present, decreased in amount. Type 2A: HMWMs absent. Type 2B: HMWMs absent (consumed). Type 2M: Normal multimer distribution. Type 2N: Normal multimer distribution. Type 3: Near-complete absence of all multimers.
What is the treatment approach in VWD? Type 1 (mild): DDAVP — releases vWF from endothelial Weibel-Palade bodies. Type 3 / severe / major surgery: vWF-containing FVIII concentrates (e.g., Humate-P) or cryoprecipitate. Type 2B: DDAVP CONTRAINDICATED; use concentrates. Adjunct for all: Tranexamic acid (antifibrinolytic) for mucosal bleeding. Women with menorrhagia: add oral contraceptives (estrogens increase vWF synthesis).
How do you differentiate VWD from Hemophilia A? VWD: autosomal (both sexes) + prolonged bleeding time + mucocutaneous bleeding + decreased vWF:Ag + decreased RIPA. Hemophilia A: X-linked recessive (males) + normal bleeding time + deep tissue bleeding + normal vWF:Ag + normal RIPA + markedly low FVIII. (Type 2N VWD mimics Hemophilia A — differentiate by autosomal history + prolonged BT + vWF:FVIII binding assay.)
How do you differentiate VWD from Bernard-Soulier Syndrome (BSS)? Both: prolonged BT + decreased RIPA + normal PT + normal aPTT. Key difference: BSS has GIANT PLATELETS + THROMBOCYTOPENIA on smear (macrothrombocytopenia) — absent in VWD. In BSS: vWF:Ag is NORMAL (defect is in platelet GpIb receptor, not vWF). In VWD: vWF:Ag is decreased.
What is Acquired Von Willebrand Syndrome and what are its causes? VWD-like syndrome in adults with NO prior bleeding history/family history. Causes: 1. Autoantibodies — lymphoproliferative disorders, myeloma, MGUS. 2. Adsorption of vWF onto tumor cells — Wilms tumor (expresses GpIb). 3. Increased shear-stress proteolysis of HMWMs — aortic stenosis (Heyde's syndrome), ventricular assist devices. 4. Hypothyroidism — decreased vWF synthesis.
A patient with longstanding aortic stenosis presents with recurrent GI bleeding. What is the mechanism? Heyde's Syndrome. High shear stress across stenotic valve → ADAMTS13-mediated proteolysis of vWF HMWMs → acquired loss of HMWMs (Type 2A-like pattern) → impaired platelet adhesion → bleeding from GI angiodysplasia. Resolves after aortic valve replacement.
What is the effect of blood group O on vWF, and what clinical mistake can it cause? Blood group O individuals have ~25% LOWER vWF levels than non-O individuals (increased glycosylation → faster clearance). This can push borderline patients below the diagnostic threshold → falsely diagnosed as Type 1 VWD. Always check ABO blood group when interpreting low vWF:Ag levels.
SUMMARY CARD — Von Willebrand Disease: Complete High-Yield Overview DEFINITION: Most common inherited bleeding disorder. Autosomal dominant (Type 3 = AR). Gene on chromosome 12. SOURCES OF vWF: Endothelium (Weibel-Palade bodies) + Platelets (alpha-granules). FUNCTIONS: 1) Platelet adhesion via GpIb. 2) Carrier/protector of Factor VIII. CLINICAL: Mucocutaneous bleeding (epistaxis, menorrhagia, bruising). Hemarthrosis only in Type 3. CLASSIC LAB: Normal platelet count + Prolonged BT/PFA-100 + Normal PT + Normal/mildly prolonged aPTT. TYPES: Type 1 = partial quantitative (75%); DDAVP works. | Type 2A = absent HMWMs, decreased RIPA, ratio <0.6. | Type 2B = hyperactive vWF, absent HMWMs, thrombocytopenia, increased low-dose RIPA, DDAVP CONTRAINDICATED. | Type 2M = decreased function, normal multimers. | Type 2N = low FVIII + normal vWF:Ag + autosomal = mimics Hemophilia A. | Type 3 = complete deficiency, AR, hemarthrosis, DDAVP ineffective. TREATMENT: DDAVP (Type 1/mild) | vWF-containing FVIII concentrate (Type 3/severe) | Tranexamic acid (adjunct) | OCP for menorrhagia. KEY DISTINCTIONS: VWD vs Hemophilia A: both sexes + prolonged BT in VWD. | VWD vs Bernard-Soulier: giant platelets + normal vWF in BSS. | VWD vs Glanzmann: RIPA normal in Glanzmann; absent ADP/collagen aggregation in Glanzmann.
Import instructions for AnkiDroid:
- Copy the entire block into a plain text file → save as
VWD_Anki.txt - Desktop Anki: File → Import → select the file → Separator: Tab → Notetype: Basic → Import
- Sync to AnkiDroid
- The
#header lines are read as settings by Anki — they do not become cards - Total: 20 clinical cards + 1 summary card = 21 cards
I also want you to give me five anki cards on von Willebrand disease diagnosis. That is, the question should be five different ways in which von Willebrand disease can be asked for exam. That is, different modes of presentation with different findings and all. Diagnosis should be, the answer should be von Willebrand disease. It should be the kind of presentations that exam question has come in five different modes of presentation. That is, they give questions like, so and so patient presented with so and so, and they found so and so findings, and then they ask questions related to that, right? First step, first step is to diagnose. So I only want the diagnosis part, five Anki cards. The answer should only be von Willebrand disease and along with the rationale for diagnosis.
#separator:tab
#html:false
#notetype:Basic
#deck:Pathology::Von Willebrand Disease
A 19-year-old female presents with heavy menstrual bleeding since menarche and frequent nosebleeds. Her mother and maternal aunt have similar complaints. Examination is normal. Lab: Platelet count 2.1 lakh (normal), PT normal, aPTT mildly prolonged, bleeding time prolonged. What is the diagnosis? Von Willebrand Disease (Type 1). RATIONALE: Autosomal dominant pattern (mother + aunt affected = both sexes, multiple generations). Mucocutaneous bleeding (epistaxis + menorrhagia) = primary hemostasis defect. Normal platelet count rules out thrombocytopenia. Normal PT rules out extrinsic pathway defect. Prolonged BT + mildly prolonged aPTT = classic VWD pattern (platelet adhesion failure + mild secondary FVIII reduction). No hemarthrosis = not pure hemophilia.
A 28-year-old male presents with excessive bleeding after a tooth extraction that required hospital admission. His younger brother had similar bleeding after a tonsillectomy. He has no spontaneous joint bleeds. Lab: Platelet count normal, PT normal, aPTT mildly prolonged, bleeding time prolonged, vWF:Ag decreased, FVIII mildly reduced. What is the diagnosis? Von Willebrand Disease. RATIONALE: Bleeding disproportionate to minor procedure (tooth extraction) with family history in a male sibling = autosomal inheritance (not X-linked). Normal platelet count + prolonged BT = primary hemostasis defect. Mildly prolonged aPTT + mildly low FVIII = secondary effect of reduced vWF carrier function. Decreased vWF:Ag confirms vWF deficiency. Absence of hemarthrosis rules against hemophilia as the primary diagnosis.
A 35-year-old woman is referred for evaluation of abnormal uterine bleeding. Investigations show: platelet count 95,000/µL (mildly low), PT normal, aPTT normal, bleeding time prolonged, vWF:Ag low, high-molecular-weight multimers absent on gel electrophoresis, and platelet aggregation occurs even at low-dose ristocetin (0.4 mg/mL). What is the diagnosis? Von Willebrand Disease Type 2B. RATIONALE: The key finding is increased/positive RIPA at LOW-dose ristocetin — this is pathognomonic of Type 2B (gain-of-function mutation → hyperactive vWF binds GpIb too avidly → spontaneous platelet consumption). This explains the MILD THROMBOCYTOPENIA (platelets consumed) and absence of HMWMs (also consumed). Most VWD types have normal platelet count — thrombocytopenia in VWD = Type 2B until proven otherwise.
A 22-year-old male is evaluated for recurrent hemarthrosis and easy bruising since childhood. His father and paternal uncle also have a bleeding disorder. Lab: Platelet count normal, PT normal, aPTT markedly prolonged, bleeding time PROLONGED, FVIII:C markedly low, vWF:Ag NORMAL, RIPA normal. What is the diagnosis? Von Willebrand Disease Type 2N (Normandy). RATIONALE: Markedly low FVIII + markedly prolonged aPTT mimics Hemophilia A. However: (1) Inheritance is AUTOSOMAL (father + uncle affected across generations, not X-linked maternal carrier pattern). (2) Bleeding time is PROLONGED — normal in Hemophilia A. (3) vWF:Ag is NORMAL — rules out Type 1/3 VWD. The combination of low FVIII + normal vWF:Ag + autosomal pattern + prolonged BT = Type 2N VWD, where the mutation is in vWF's FVIII-binding domain → unprotected FVIII is rapidly degraded.
A 10-year-old boy presents with spontaneous gum bleeding, recurrent epistaxis, and a recent episode of knee joint swelling (hemarthrosis). His parents are consanguineous. Lab: Platelet count normal, PT normal, aPTT markedly prolonged, bleeding time markedly prolonged, vWF:Ag nearly absent, FVIII:C markedly low. What is the diagnosis? Von Willebrand Disease Type 3. RATIONALE: Consanguinity = autosomal recessive inheritance. Near-complete absence of vWF → (1) Severely impaired platelet adhesion → markedly prolonged BT + mucocutaneous bleeding. (2) No vWF to carry FVIII → FVIII rapidly degraded → markedly low FVIII → prolonged aPTT + hemarthrosis (mimicking Hemophilia A). Type 3 is the ONLY VWD type that causes hemarthrosis — because it is the only type with severe enough FVIII deficiency. The combination of hemarthrosis + prolonged BT + consanguinity + near-absent vWF:Ag clinches Type 3 VWD.
| Card | Presentation Mode | Key Trap |
|---|---|---|
| 1 | Adolescent female, menorrhagia + epistaxis, family history | Recognizing autosomal pattern + mucocutaneous = VWD |
| 2 | Male with post-procedural bleed, family history in brother | Male patient + autosomal = not hemophilia |
| 3 | Thrombocytopenia + absent HMWMs + low-dose RIPA positive | Thrombocytopenia in VWD = always Type 2B |
| 4 | Looks exactly like Hemophilia A but autosomal + prolonged BT | Differentiating Type 2N from Hemophilia A |
| 5 | Consanguinity + hemarthrosis + near-absent vWF | Only Type 3 causes hemarthrosis in VWD |