Explain me a concept of explain me a concept of myocardial infraction myocardial infraction in fractioon
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive material from multiple authoritative textbooks. Here is the full, detailed explanation:
Myocardial Infarction (MI) - "Heart Attack"
What Is It?
Myocardial infarction (MI) is necrosis (death) of heart muscle resulting from ischemia - inadequate blood supply to meet the metabolic demand of the myocardium. The 2018 joint task force of U.S. and European Cardiology groups defines it as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
It is commonly called a "heart attack" and is one of the leading causes of death worldwide. - Robbins & Kumar Basic Pathology, p. 353
Causes and Risk Factors
The vast majority (~90%) of MIs result from acute thrombosis of a coronary artery overlying a disrupted atherosclerotic plaque. The remaining ~10% occur from:
- Coronary artery vasospasm
- Embolism from mural thrombi (e.g., in atrial fibrillation) or valve vegetations
- Severe fixed coronary atherosclerosis without thrombosis (especially subendocardial infarcts)
- Disorders of small intramyocardial arterioles: vasculitis, amyloid deposition, sickle cell disease
Key risk factors: aging, male sex (women are relatively protected until menopause), hypertension, diabetes, dyslipidemia, smoking, and family history. About 10% of MIs occur before age 40. - Robbins & Kumar Basic Pathology, p. 353
Pathogenesis - How Does MI Happen?
The typical sequence of events:
-
An atheromatous plaque erodes or ruptures due to endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to circulating blood.
-
Platelets adhere, aggregate, and activate, releasing thromboxane A2, ADP, and serotonin - causing more platelet aggregation and vasospasm.
-
Coagulation is activated by exposed tissue factor, adding to the growing thrombus.
-
Within minutes, the enlarging thrombus completely occludes the coronary artery lumen.
Angiography performed within 4 hours of onset shows coronary thrombosis in almost 90% of cases. - Robbins & Kumar Basic Pathology, p. 354
Histology of coronary atherosclerosis and thrombosis:
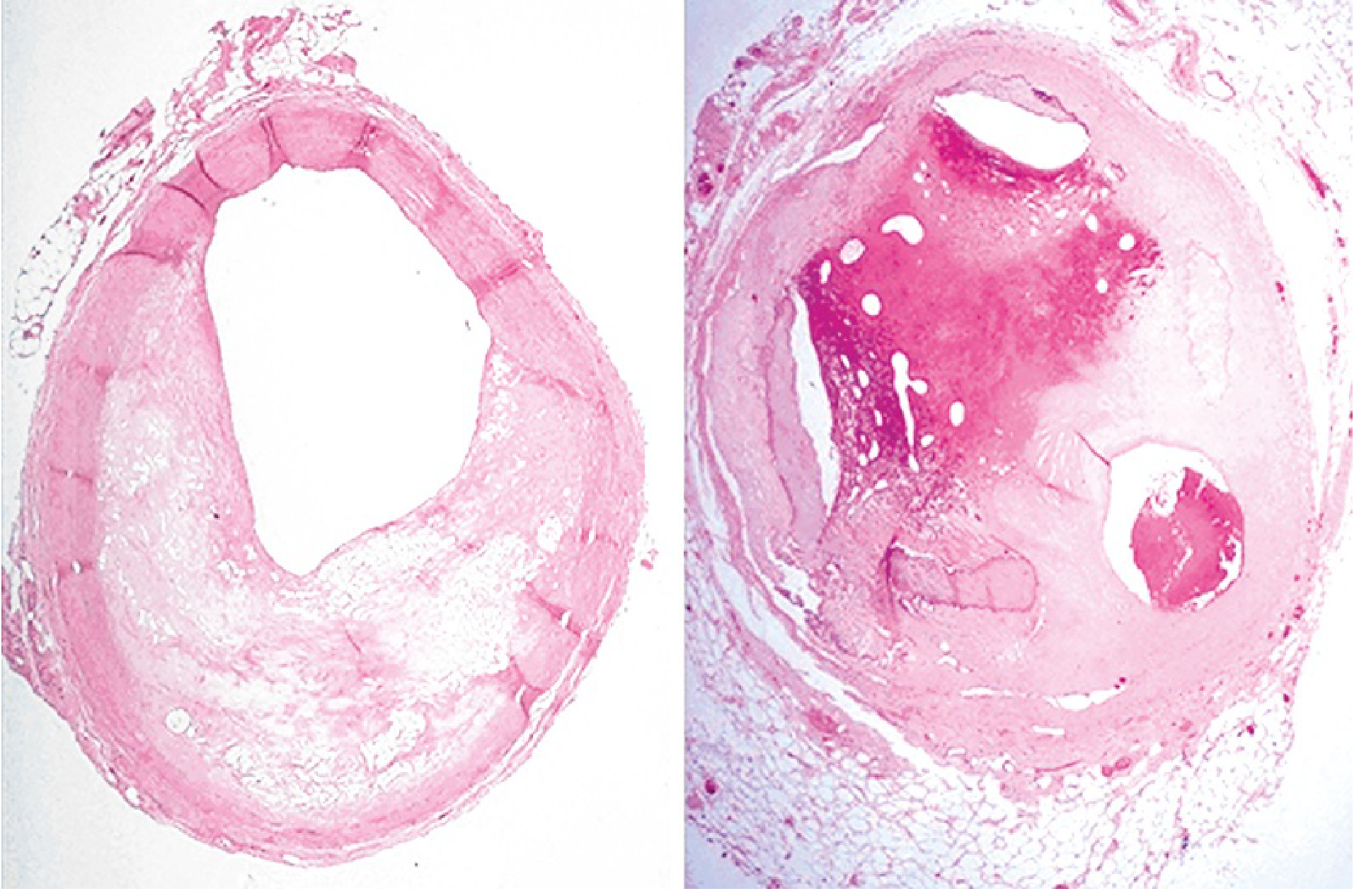
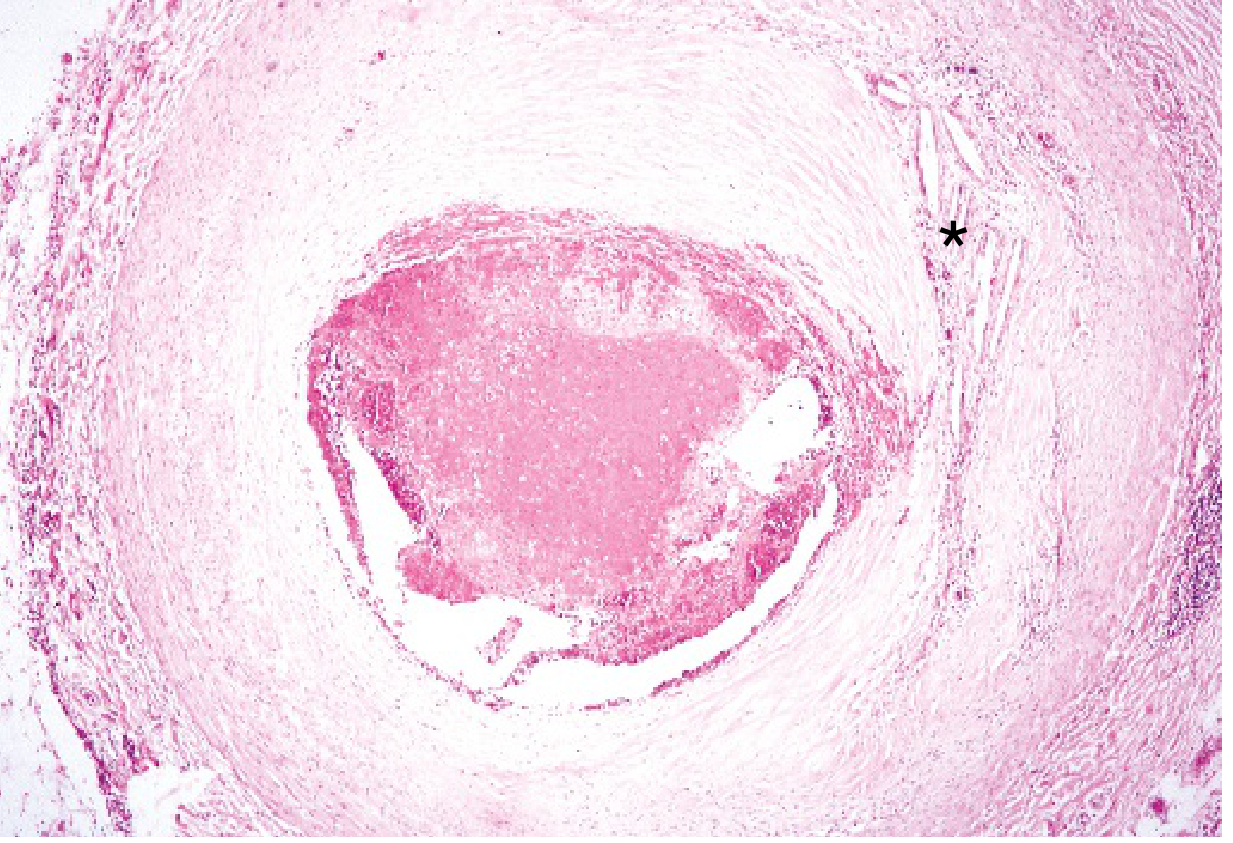
What Happens to the Myocardium After Occlusion
Immediate metabolic changes (seconds to minutes)
- Aerobic metabolism ceases within seconds of obstruction
- ATP drops rapidly; lactic acid and other toxic metabolites accumulate
- Contractility is lost within minutes
- These changes are reversible at this stage
Irreversible injury threshold (20-40 minutes)
- If ischemia persists for 20-40 minutes, irreversible coagulative necrosis of myocytes occurs
- The sarcolemmal membrane ruptures, leaking intracellular macromolecules (troponins, CK-MB) into the bloodstream - this is the basis of cardiac biomarker testing
Wavefront of necrosis
Irreversible injury starts in the subendocardial zone first (it is most distal from epicardial vessels and under highest intramural pressure). With prolonged ischemia, the wavefront moves outward toward the epicardium, driven by tissue edema and reactive oxygen species.
Oxygen requirements
The heart requires about 1.3 mL O2/100g of muscle/min just to survive. The normal resting left ventricle receives ~8 mL O2/100g/min. Even 15-30% of normal coronary flow may be enough to keep muscle alive. In the core of a large infarct with near-zero collateral flow, muscle death is inevitable. - Guyton & Hall Medical Physiology, p. 271
Types of MI
| Type | Description |
|---|---|
| STEMI (ST-elevation MI) | Full-thickness (transmural) infarct; complete occlusion |
| NSTEMI (Non-ST-elevation MI) | Partial-thickness (subendocardial); partial or transient occlusion |
| Type 1 | Spontaneous MI from plaque rupture/erosion |
| Type 2 | MI secondary to supply-demand mismatch (e.g., severe anemia, tachycardia) |
| Types 3-5 | MI related to cardiac death, PCI, or CABG procedures |
(Fourth Universal Definition of MI, 2018)
ECG Changes in MI
The three major electrical abnormalities in acute MI are:
| Defect in Infarcted Cells | Current Flow | ECG Change Over Infarct |
|---|---|---|
| Rapid repolarization (accelerated K+ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential (loss of intracellular K+) | Into infarct | TQ depression (appears as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
The hallmark of acute MI is ST-segment elevation in leads overlying the infarcted area. Leads on the opposite side show ST depression (reciprocal changes).
After days to weeks, ST changes resolve. The dead, electrically silent tissue manifests as pathological Q waves - because the infarct area fails to contribute positive voltage during depolarization. - Ganong's Review of Medical Physiology, p. 534
Causes of Death After MI
1. Decreased Cardiac Output - Cardiogenic Shock
When healthy portions of the ventricle contract, the infarcted, non-functional muscle bulges outward instead of contracting inward - this is called "systolic stretch" (paradoxical wall motion). This wastes pumping force and dramatically reduces cardiac output.
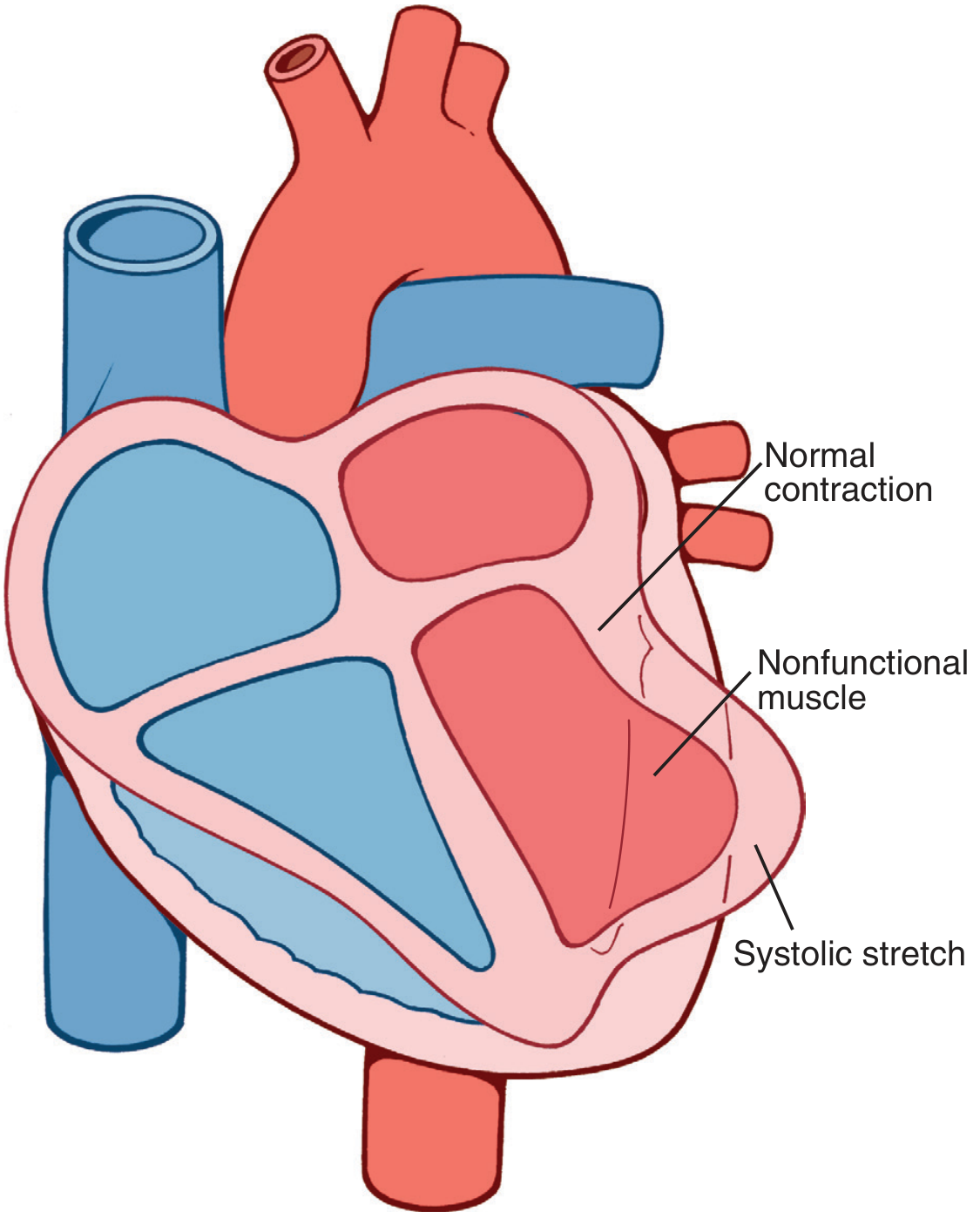
Cardiogenic shock occurs when >40% of the left ventricle is infarcted. Mortality is 40-50% even with modern treatment.
2. Pulmonary Edema
Failure to pump blood forward causes damming of blood in the pulmonary circulation. Reduced cardiac output impairs renal perfusion and urine output, gradually increasing blood volume - leading to acute pulmonary edema, sometimes appearing days after the initial event.
3. Ventricular Fibrillation
In 80-90% of cardiac deaths after MI, death is from ventricular fibrillation (not mechanical failure). Factors promoting fibrillation:
- K+ leaks out of ischemic cells → elevated extracellular K+ → increased irritability
- Injury currents from partially repolarized ischemic muscle
- Sympathetic activation (baroreceptor reflex response to low blood pressure) increases cardiac irritability
- Local acidosis from lactic acid
Two peak periods for fibrillation: within 10 minutes of infarction, and again at ~1 hour. - Guyton & Hall Medical Physiology, p. 272
4. Cardiac Rupture (less common)
Necrotic myocardium can rupture, causing cardiac tamponade or acute mitral regurgitation from papillary muscle rupture.
Stunned vs. Hibernating Myocardium
- Stunned myocardium: When reperfusion occurs before irreversible injury, the myocardium is saved but remains non-contractile for days due to persistent biochemical abnormalities. This is reversible.
- Hibernating myocardium: Chronically hypoperfused but viable muscle that reduces its function to match supply. Contractility recovers with revascularization.
Cardiac Biomarkers
Sarcolemmal rupture in necrotic myocytes releases intracellular proteins into the bloodstream:
| Biomarker | Rises | Peaks | Returns to Normal |
|---|---|---|---|
| Troponin I/T (most specific) | 2-4 hrs | 24-48 hrs | 5-14 days |
| CK-MB | 4-6 hrs | 12-24 hrs | 48-72 hrs |
| Myoglobin | 1-2 hrs | 6-9 hrs | 24 hrs |
Troponin is the gold-standard biomarker because it is highly sensitive and cardiac-specific.
Treatment Principles
The goal is to restore perfusion as quickly as possible (time = muscle):
- Primary PCI (percutaneous coronary intervention / angioplasty + stent): treatment of choice for STEMI, ideally within 90 minutes of first medical contact
- Thrombolysis: if PCI is not available within 120 minutes
- Antiplatelet therapy: aspirin + P2Y12 inhibitor (clopidogrel, ticagrelor)
- Anticoagulation: heparin
- Beta-blockers: reduce cardiac work and arrhythmia risk
- ACE inhibitors / ARBs: reduce remodeling post-MI
- Statins: stabilize atherosclerotic plaques long-term
Summary in one sentence: MI is death of heart muscle due to prolonged ischemia, most often from coronary artery thrombosis on a ruptured atherosclerotic plaque, causing a cascade from ATP depletion to coagulative necrosis, manifesting clinically as chest pain, ECG changes, and rising cardiac biomarkers - and potentially leading to arrhythmia, pump failure, or death if not treated urgently.
Sources: Robbins & Kumar Basic Pathology; Guyton & Hall Textbook of Medical Physiology; Ganong's Review of Medical Physiology; Braunwald's Heart Disease
This is a shared conversation. Sign in to Orris to start your own chat.