Disorders of sexual development in detail from standard textbooks for post graduate level with definitions clinical features pathophysiology investigations and it's management of all 46xx and 46xy disorders of sexual development
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Searching PubMed
"disorders of sexual development" classification management
I now have comprehensive textbook data from Campbell-Walsh Urology, Smith & Tanagho's General Urology, and Harrison's Principles of Internal Medicine. I have enough material to write a thorough postgraduate-level answer. Let me now compose the full detailed response.
Disorders of Sexual Development (DSD) - A Postgraduate Reference
1. DEFINITION AND CLASSIFICATION
Disorders of Sexual Development (DSD) are congenital conditions in which chromosomal, gonadal, or anatomical sex is atypical. The term DSD replaced the older terminology of "hermaphroditism" and "pseudohermaphroditism" following the Chicago Consensus Conference (2006), which recognized these terms as stigmatizing.
"DSDs are congenital diseases in which the chromosomal, gonadal, or anatomic sex is abnormal and embraces a wide-ranging spectrum of phenotypes classified in five different groups." - Campbell-Walsh-Wein Urology
2006 Chicago Consensus Classification:
| Category | Old Terminology | Example |
|---|---|---|
| Sex chromosome DSD | Klinefelter, Turner | 45,X; 47,XXY |
| 46,XX DSD | Female pseudohermaphroditism | CAH, maternal androgens |
| 46,XY DSD | Male pseudohermaphroditism | AIS, 5a-reductase deficiency |
| Ovotesticular DSD | True hermaphroditism | Both ovarian + testicular tissue |
| Gonadal dysgenesis | Streak gonads | Swyer syndrome |
The current nomenclature includes karyotype + clinical descriptor + molecular basis when known. For example: "46,XY DSD, complete gonadal dysgenesis with SF1 mutation" (Campbell-Walsh-Wein Urology).
2. NORMAL SEXUAL DIFFERENTIATION (Embryological Basis)
Understanding DSD requires knowledge of the three sequential steps of sexual differentiation:
Step 1: Chromosomal Sex (Fertilization)
- XX = genetic female; XY = genetic male
- The SRY gene (Sex-determining Region on Y chromosome, located on the short arm of Y) is the master switch for testicular development
- SRY activates a cascade involving SOX-9, SF-1, WT-1, FTZ-F1, and LIM-1 which direct testis formation
- In XX females, two copies of DAX-1 (on Xp) suppress StAR protein and inhibit testicular development, defaulting to ovarian development
Step 2: Gonadal Sex (Weeks 6-7)
- Gonads arise from urogenital ridges (week 4) - bipotential until week 6-7
- Primordial germ cells migrate from yolk sac endoderm
- In XY: SRY directs Sertoli cell differentiation → Sertoli cells produce AMH (Anti-Mullerian Hormone) → Müllerian regression; Leydig cells produce testosterone → Wolffian duct development
- In XX: Ovarian development is the "default" pathway; granulosa cells form, Müllerian ducts persist (form uterus, fallopian tubes, upper vagina)
Step 3: Phenotypic Sex (Weeks 8-12)
| Hormone | Source | Action |
|---|---|---|
| Testosterone | Fetal Leydig cells | Wolffian duct development → epididymis, vas deferens, seminal vesicles |
| DHT (from T via 5a-reductase type 2) | External genitalia tissue | External male genitalia formation (scrotum, penis, prostate) |
| AMH | Fetal Sertoli cells | Müllerian duct regression |
| Absence of androgens | - | Female external genitalia (default) |
3. WHEN DSD IS SUSPECTED
Patients with DSD present in four main scenarios (Smith & Tanagho's General Urology):
- Newborn period: atypical/ambiguous genitalia, or discordant phenotype from prenatal karyotype
- Inappropriate pubertal development: virilization in a genetic female, or feminization in a genetic male
- Delayed puberty: primary amenorrhea or absent pubertal changes
- Later in life: infertility, inguinal hernia containing testis in a phenotypic female
4. INITIAL CLINICAL EVALUATION
History
- Family history of unexplained infant deaths, infertility, amenorrhea, hirsutism
- Drug ingestion during pregnancy (progestogens, androgens, finasteride, danazol, stilbestrol - see table below)
- Maternal virilization during pregnancy (suggests virilizing tumor)
- Consanguinity (autosomal recessive disorders)
Drugs causing DSD if taken during pregnancy:
- C21-steroid medroxyprogesterone acetate (progesterone)
- Finasteride, Leuprolide acetate
- Stilbestrol, Danazol, Norethynodrel, Ethisterone, Norethindrone
Physical Examination
- Palpate gonads: a gonad in the inguinal canal or labioscrotal fold is almost always a testis (ovaries do not descend) - this virtually excludes 46,XX DSD
- Assess phallus size: stretched penile length < 2.5 cm = micropenis; clitoromegaly = > 1 cm
- Labioscrotal fusion: degree (partial vs. complete) and pigmentation
- Urogenital sinus: presence suggests androgen excess in 46,XX
- Midline structures: palpate for uterus abdominally and rectally
- Stigmata: web neck, shield chest, cardiac murmurs (Turner), gynecomastia (Klinefelter)
- Prader staging for degree of virilization in 46,XX DSD (I-V)
Investigations
| Test | Purpose |
|---|---|
| Karyotype (peripheral blood) | First-line, defines chromosomal sex |
| Serum 17-OHP | Elevated in 21-hydroxylase deficiency CAH |
| Serum electrolytes | Hyponatremia/hyperkalemia in salt-wasting CAH |
| Serum glucose | Hypoglycemia in salt-wasting |
| LH, FSH | Distinguish primary vs. secondary hypogonadism |
| Testosterone, DHT, T/DHT ratio | Elevated T:DHT ratio (>30:1) suggests 5a-reductase deficiency |
| Testosterone precursors (after hCG stimulation) | Identify specific enzyme defects |
| hCG stimulation test | If testosterone response < 2 ng/mL → no functional testicular tissue; >2 ng/mL → testosterone synthesis or action defect |
| Androgen receptor binding assay | Genital skin fibroblast culture - confirms AIS |
| AMH (Müllerian Inhibiting Substance) | Detectable in AIS (testes present), undetectable in streak gonads |
| Pelvic ultrasound | Müllerian structures (uterus, ovaries), gonad location |
| Genitography/vaginoscopy | Define urogenital sinus anatomy |
| MRI pelvis | Internal anatomy, gonad localization |
| Adrenal ultrasound/CT | Enlarged adrenals in CAH; adrenal tumors |
| Diagnostic laparoscopy/laparotomy | Gonad biopsy when sex assignment depends on histology; removal of dysgenetic gonads |
| Molecular genetics | SRY, CYP21A2, AR, SRD5A2, AMH, SF1 mutations |
5. 46,XX DSD (Masculinized Female / Former Female Pseudohermaphroditism)
"46,XX DSD is a disorder of phenotypic sexual development in which 46,XX individuals with ovaries have partially masculinized phenotype and ambiguous genitalia." - Campbell-Walsh-Wein Urology
The gonads are ovaries, the internal genitalia are Müllerian structures (uterus and fallopian tubes are present), but the external genitalia are virilized. The underlying mechanism is excess androgens during fetal development.
Causes of 46,XX DSD:
5.1 Congenital Adrenal Hyperplasia (CAH) - Most Common Cause
Definition: A group of autosomal recessive disorders caused by enzyme defects in the cortisol biosynthetic pathway, leading to ACTH hypersecretion, adrenal hyperplasia, and excess androgen production.
Genetics: Autosomal recessive. The CYP21A2 gene is on chromosome 6p21.3 within the HLA complex. A pseudogene (CYP21PA1) is 98% homologous and accounts for most mutations (gene conversion or deletion events during meiosis).
Enzyme Defects in the Cortisol Pathway:
Cholesterol
↓ StAR → P450scc
Δ5-Pregnenolone → 17α-OH Pregnenolone → DHEA
↓ 3β-HSD ↓ 3β-HSD
Progesterone → 17α-OH Progesterone → Androstenedione → Testosterone
↓ 21-OH ↓ 21-OH
11-DOC DOC → Corticosterone → Aldosterone
↓ 11β-OH
Cortisol
5.1a: 21-Hydroxylase Deficiency (95% of all CAH)
Pathophysiology:
- Block in cortisol synthesis → ↑ ACTH → adrenal hyperplasia → ↑ 17-OHP → shunted to androgens (androstenedione → testosterone)
- Aldosterone synthesis also impaired in severe form → salt wasting
Clinical Forms:
| Form | Features |
|---|---|
| Classic salt-wasting (75%) | Virilization + aldosterone deficiency → hyponatremia, hyperkalemia, dehydration, shock in first 2-3 weeks of life; potentially lethal if unrecognized |
| Classic simple virilizing (25%) | Virilization without salt wasting; adequate mineralocorticoid synthesis |
| Non-classic (late-onset) | No virilization or salt wasting; presents with premature adrenarche, hirsutism, acne, oligomenorrhea, infertility in females |
Clinical Features in 46,XX (Female):
- At birth: ambiguous genitalia - clitoromegaly, labioscrotal fusion, urogenital sinus
- Prader grading I-V (I = mild clitoromegaly; V = complete penile urethra, fused scrotum)
- Normal Müllerian structures (uterus, ovaries intact)
- Salt-wasting crisis at 1-3 weeks of life (feeding difficulties, vomiting, dehydration, hyperpigmentation from ACTH excess)
- Without treatment: progressive virilization, advanced bone age, short final height
Clinical Features in 46,XY (Male):
- Genital phenotype normal at birth
- Salt-wasting crisis if severe
- Precocious pseudo-puberty (pubic hair, penile growth, accelerated growth, advanced bone age but testes remain prepubertal)
- Testicular adrenal rest tumors (TART) in adulthood - can cause infertility
Investigations:
- Serum 17-OHP: markedly elevated (>100 ng/mL in classic; 10-100 in non-classic); the definitive diagnostic marker
- Serum electrolytes: Na↓, K↑ (salt-wasting)
- Plasma renin activity: elevated in salt-wasting
- Androstenedione, DHEAS: elevated
- Karyotype: 46,XX
- Pelvic ultrasound: normal Müllerian structures, adrenal enlargement
- Neonatal screening: 17-OHP heel-prick test (day 2-4 of life) - now standard in many countries
- ACTH stimulation test for non-classic: 17-OHP >10 ng/mL at 60 min is diagnostic
- HLA typing (6p21.3 haplotype)
- CYP21A2 gene mutation analysis
Management:
Medical:
- Glucocorticoid replacement: Hydrocortisone 10-15 mg/m²/day in 3 divided doses (preferred in children to minimize growth suppression); prednisolone or dexamethasone in adults
- Mineralocorticoid replacement (salt-wasting): Fludrocortisone 0.05-0.2 mg/day + sodium chloride supplementation (1-2 g/day) in infants
- Monitoring: 17-OHP, androstenedione, plasma renin activity, growth velocity, bone age
- Stress dosing: 2-3x maintenance dose during illness, surgery, trauma ("stress rule")
- Prenatal treatment (experimental/controversial): Dexamethasone 20 µg/kg/day started before 8 weeks gestation to prevent virilization of affected female fetus (must diagnose early via CVS)
Surgical:
- Indicated for Prader III-V degree of virilization
- Timing: traditionally at 2-6 months of age (clitoroplasty, vaginoplasty); modern consensus favors deferring vaginoplasty to adolescence
- Clitoroplasty: nerve-sparing reduction; never total clitorectomy (preserves sensation and sexual function)
- Vaginoplasty: flap vaginoplasty or pull-through procedure
- Ongoing psychosocial support and gender identity counseling
5.1b: 11β-Hydroxylase Deficiency (5-8% of CAH)
Pathophysiology:
- Block at 11β-hydroxylase (CYP11B1) → ↑ 11-deoxycortisol (compound S) and 11-deoxycorticosterone (DOC) → DOC has mineralocorticoid activity → hypertension
- Androgenic pathway is intact → virilization
Clinical Features:
- Virilization of 46,XX female (ambiguous genitalia at birth)
- Hypertension (characteristic feature, not seen in 21-OH deficiency)
- Hypokalemia (from DOC excess)
- Low aldosterone and renin (suppressed by DOC)
- No salt wasting (DOC provides mineralocorticoid effect)
Investigations:
- Elevated 11-deoxycortisol and 11-DOC
- Elevated 17-OHP (modest, less than 21-OH deficiency)
- Elevated DHEAS, androstenedione
- Low renin activity
Management:
- Glucocorticoid replacement (suppresses DOC → corrects hypertension)
- Surgical reconstruction as needed
- No mineralocorticoid supplementation needed
5.1c: 3β-Hydroxysteroid Dehydrogenase (3β-HSD) Deficiency
Pathophysiology:
- Block in conversion of Δ5 to Δ4 steroids → accumulation of DHEA, pregnenolone
- DHEA is a weak androgen → mild virilization in 46,XX females
- Severe salt wasting because both cortisol and aldosterone synthesis are impaired
Clinical Features in 46,XX:
- Mild virilization (clitoromegaly) - less severe than 21-OH deficiency
- Severe salt-wasting crisis
- Elevated DHEA/DHEAS, low cortisol, low aldosterone
Clinical Features in 46,XY:
- Paradoxically - undervirilization (ambiguous genitalia) because testosterone synthesis requires 3β-HSD
- Salt wasting
Management: Glucocorticoid + mineralocorticoid replacement; surgical correction as needed
5.1d: 17α-Hydroxylase Deficiency
Pathophysiology:
- CYP17A1 enzyme deficiency
- In 46,XX: no sex steroid production → primary amenorrhea, sexual infantilism, no virilization
- Excess mineralocorticoids (DOC, corticosterone) → hypertension, hypokalemia
Clinical Features in 46,XX:
- Normal or female-appearing external genitalia at birth
- Primary amenorrhea, sexual infantilism at puberty
- Hypertension, hypokalemia
Clinical Features in 46,XY:
- Female or ambiguous external genitalia (no testosterone synthesis)
- See Section 6 (46,XY DSD) for details
5.2 Maternal Androgen Excess (Exogenous/Endogenous)
Exogenous: Maternal ingestion of androgens or progestogens during pregnancy (see drug table above)
Endogenous: Virilizing ovarian or adrenal tumors in the mother (rare) - Sertoli-Leydig cell tumor, luteoma of pregnancy
Features:
- 46,XX infant, normal Müllerian structures, normal ovaries
- Virilized external genitalia only (not progressive)
- Spontaneous improvement after delivery as maternal hormones clear
- No adrenal insufficiency
Management: No specific treatment; surgical correction if needed; prognosis excellent
5.3 Ovotesticular DSD (Former True Hermaphroditism)
Definition: Coexistence of both ovarian tissue (with follicles) and testicular tissue (with seminiferous tubules) in the same individual, in the same gonad (ovotestis) or separately.
Karyotype: Most commonly 46,XX (~60%), but also 46,XY (~10%) or mosaics (45,X/46,XY or 46,XX/46,XY ~30%)
Pathophysiology:
- In 46,XX cases: cryptic Y-DNA (SRY translocation to X chromosome or autosome), or SRY-independent gonadal sex reversal
- Gonads may be an ovotestis, or separate ovary on one side and testis on the other
Clinical Features:
- Variable - ambiguous genitalia at birth most common
- Uterus usually present (can vary)
- 70% raised as males
- Breast development and menstruation possible at puberty
- Inguinal hernia in 50%
Diagnosis: Gonad biopsy (laparoscopic) showing both ovarian follicles and testicular tubules
Management:
- Gender assignment based on gonadal anatomy, potential for function, and family/cultural factors
- Remove discordant gonadal tissue
- Gonadal cancer risk: ~4% (lower than gonadal dysgenesis)
6. 46,XY DSD (Undervirilized Male / Former Male Pseudohermaphroditism)
In 46,XY DSD, the karyotype is male (46,XY), testes are usually present, but external genitalia are incompletely virilized ranging from female-appearing to ambiguous.
The fundamental defect is failure of androgen production or action (or rarely, failure of AMH action).
Refer to the diagnostic algorithm above (Fig. 43-14, Smith & Tanagho) - the key branch point is whether Müllerian structures are present or absent, and the testosterone response to hCG stimulation.
6.1 Disorders of Androgen Synthesis
6.1a: StAR Protein Deficiency (Congenital Lipoid Adrenal Hyperplasia)
Pathophysiology:
- StAR (Steroidogenic Acute Regulatory protein) transports cholesterol across the mitochondrial membrane - the first and rate-limiting step in steroidogenesis
- Complete block → no sex steroids, no glucocorticoids, no mineralocorticoids
- Most severe form of adrenal hyperplasia
Clinical Features:
- 46,XY: female or ambiguous external genitalia (no testosterone or DHT)
- 46,XX: normal female genitalia
- Both: severe salt-wasting crisis in neonatal period (life-threatening)
- Massive adrenal enlargement (lipid-laden adrenal cortex on imaging)
- Primary adrenal insufficiency
Management: Lifelong glucocorticoid + mineralocorticoid replacement; gender assignment toward female in severely undervirilized 46,XY; gonadectomy
6.1b: 3β-HSD Deficiency in 46,XY
Already discussed above. Results in undervirilization (ambiguous genitalia) because testosterone requires 3β-HSD for synthesis.
6.1c: 17α-Hydroxylase / 17,20-Lyase Deficiency in 46,XY
Pathophysiology:
- CYP17A1 encodes both 17α-hydroxylase and 17,20-lyase activities
- Deficiency → no sex steroid synthesis (no DHEA, no androstenedione, no testosterone)
- Excess DOC and corticosterone → hypertension, hypokalemia
Clinical Features in 46,XY:
- Female or ambiguous external genitalia
- No Müllerian structures (AMH from testes is normal)
- Absent pubertal development
- Hypertension and hypokalemia
- Elevated progesterone and pregnenolone; low cortisol
Management: Glucocorticoid therapy (corrects hypertension by suppressing ACTH and DOC); sex steroid replacement at puberty; sex assignment toward female in complete cases
6.1d: 17β-Hydroxysteroid Dehydrogenase (17β-HSD) Deficiency
Pathophysiology:
- 17β-HSD converts androstenedione to testosterone (and estrone to estradiol)
- Deficiency → cannot synthesize testosterone from androstenedione
- Autosomal recessive; gene HSD17B3 on chromosome 9q22
Clinical Features in 46,XY:
- Female-appearing external genitalia at birth (no scrotum, no penis; testes often undescended)
- No Müllerian structures (AMH normal - Müllerian regression occurs)
- At puberty: virilization occurs (androstenedione peripherally converted to testosterone by other isoenzymes of 17β-HSD)
- Clitoral/penile enlargement, virilization, often with gender role change to male
- Elevated androstenedione/testosterone ratio (>3:1) is diagnostic
Investigations: hCG stimulation → elevated androstenedione, low testosterone; elevated A:T ratio; HSD17B3 gene mutation analysis
Management:
- If raised female: gonadectomy before puberty, estrogen replacement at puberty
- If male identity: testosterone therapy, surgical correction of genitalia
6.2 5α-Reductase Type 2 Deficiency
"5α-Reductase type 2 deficiency is an autosomal recessive transmitted disorder affecting the formation of the male genitalia." - Smith & Tanagho's General Urology
Pathophysiology:
- SRD5A2 gene mutation → failure to convert testosterone (T) to dihydrotestosterone (DHT)
- DHT is 5-10x more potent than T and is essential for external genitalia formation
- Type 2 5α-reductase predominates in external genitalia and prostate; type 1 in skin and nongenital tissues
- DHT is localized to the midline urethral seam during fetal development; failure of seam fusion → hypospadias
Clinical Features:
- At birth: small phallus, severe hypospadias, bifid scrotum, blind vaginal pouch / residual prostatic utricle
- Testes often undescended (inguinal)
- Müllerian structures absent (AMH normal)
- Wolffian structures present (epididymis, vas deferens)
- At puberty: significant virilization - phallic enlargement, scrotal development, voice change (testosterone levels surge and overwhelm the deficiency via type 1 enzyme or receptor saturation)
- Gender identity: predominantly male despite female-rearing in many cultures. In Dominican Republic (high prevalence), the condition was called "guevedoces" (testicles at twelve) - most children change gender identity from female to male at puberty
Diagnosis:
- T:DHT ratio > 30:1 (normally < 10:1) after hCG stimulation
- Elevated testosterone, low DHT
- SRD5A2 gene mutation analysis
- Normal karyotype (46,XY)
- Normal AMH
Management:
- Male sex assignment: testosterone or DHT therapy; hypospadias repair, orchiopexy
- Female sex assignment: gonadectomy before puberty, estrogen replacement; vaginal reconstruction
- Pregnancies reported via ICSI (preserved spermatogenesis in some cases)
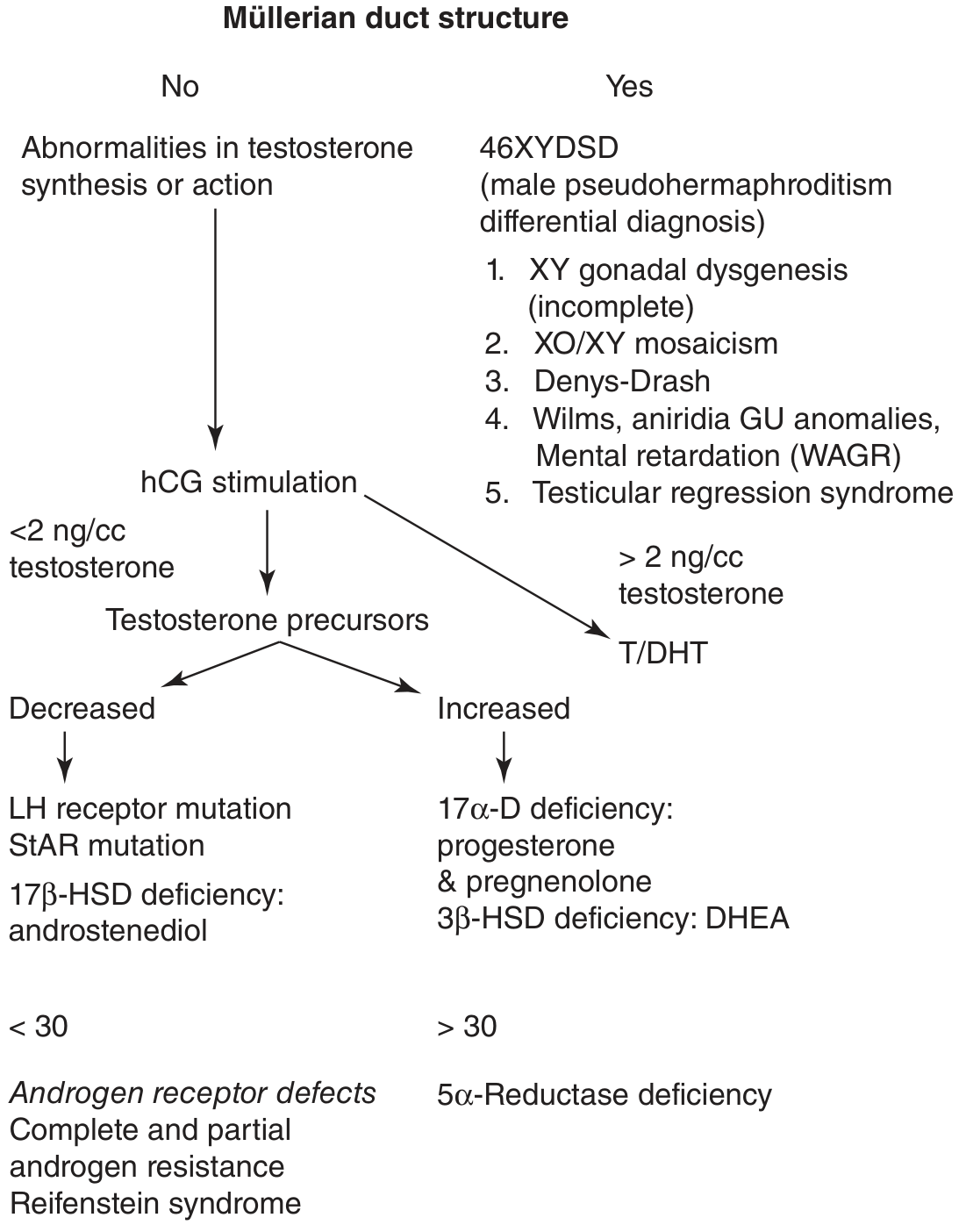
6.3 Androgen Insensitivity Syndrome (AIS)
Definition: X-linked disorder caused by loss-of-function mutations in the androgen receptor (AR) gene (chromosome Xq11-12), resulting in complete or partial resistance to androgens despite normal or elevated testosterone levels.
"Androgen resistance ranges from partial to complete owing to a defect in the AR. Patients with complete androgen resistance have a 46,XY karyotype but have unambiguous female external genitalia, hypoplastic labia majora, a blind vaginal pouch, and an absent uterus." - Smith & Tanagho's General Urology
Genetics: X-linked recessive; AR gene on Xq11-12; >1000 mutations identified; sporadic mutations in 30-40% of cases
Pathophysiology:
- Testes develop normally (SRY intact), AMH produced normally → Müllerian regression (no uterus, no tubes)
- Testosterone is produced normally or in excess (due to LH elevation)
- However, testosterone/DHT cannot act on target tissues (AR mutation) → no male differentiation of external genitalia
- Peripheral aromatization of excess testosterone to estradiol → breast development (via ER, which is functional)
- Brain "masculinization" also fails (functional AR required)
6.3a: Complete AIS (CAIS) - "Testicular Feminization"
Clinical Features:
- Unambiguous female external genitalia
- Blind vaginal pouch (shortened vagina, no cervix, no uterus)
- Absent or sparse axillary and pubic hair (androgens drive hair growth)
- Normal breast development at puberty
- Primary amenorrhea - most common presentation in adolescence
- Testes may be in abdomen, inguinal canal (presents as inguinal hernia in ~1% of prepubertal females undergoing hernia repair - a classic exam question), or labia majora
- Gender identity: female (brain "masculinization" absent)
Investigations:
- Karyotype: 46,XY in a phenotypic female
- Serum testosterone: elevated (male range or above)
- LH: elevated (loss of androgen feedback on hypothalamus-pituitary)
- FSH: normal or mildly elevated
- AMH: detectable (confirms testes present)
- Serum estradiol: elevated (aromatization from excess T)
- Pelvic ultrasound/MRI: absent uterus, blind vaginal pouch; testes located intra-abdominally or inguinally
- Androgen receptor binding assay (fibroblast culture): absent or decreased binding
- AR gene sequencing: confirms mutation
Management:
- Gonadectomy: the gonads carry a 10% risk of malignancy post-puberty (most commonly dysgerminoma/seminoma). Options:
- Delay gonadectomy until after puberty (allows natural breast development, bone density accrual), then gonadectomy + estrogen replacement
- Early gonadectomy (at diagnosis) + exogenous estrogen at puberty
- Current preference: most centers defer until after puberty
- Hormone replacement: estrogen replacement lifelong after gonadectomy (maintains bone density, cardiovascular health)
- Vaginal augmentation: if vaginal length is inadequate; self-dilation (Frank technique) preferred first-line; surgical vaginoplasty (McIndoe procedure) if dilation fails
- Psychological support: disclosure is critical - full open communication with the patient about their diagnosis is now the standard approach
- Fertility: no fertility possible (no uterus); sperm retrieval not possible (testes are dysgenetic)
6.3b: Partial AIS (PAIS) - Reifenstein Syndrome and Others
Clinical Features:
- Wide spectrum: from mild hypospadias with gynecomastia to micropenis with cryptorchidism to severe ambiguous genitalia with clitoromegaly
- Classic Reifenstein syndrome: hypospadias, cryptorchidism, gynecomastia, infertility
- No Müllerian structures (AMH present)
- Virilizes partially at puberty
Investigations: Same as CAIS; AR gene sequencing shows partial loss-of-function mutation
Management:
- Sex assignment is complex and individualized
- Male assignment: orchiopexy, hypospadias repair, testosterone therapy; gynecomastia surgery
- Female assignment: gonadectomy, estrogen replacement, vaginal reconstruction
- The decision depends on degree of virilization, AR function, potential for erectile function, and psychosocial factors
6.4 XY Complete Gonadal Dysgenesis (Swyer Syndrome)
Definition: 46,XY individuals with streak gonads (fibrous bands without functional gonadal tissue), female phenotype, and absent Müllerian regression (Müllerian structures present because AMH is not produced).
Pathophysiology:
- Mutations in SRY (15-20%), SOX9, SF1, DHH, or unknown factors → failure of testicular differentiation → no testosterone, no AMH production
- No androgens → female external genitalia
- No AMH → Müllerian structures present (uterus, tubes)
- Streak gonads cannot sustain ovarian function
Clinical Features:
- Normal female external genitalia
- Uterus and fallopian tubes present (Müllerian structures intact)
- Primary amenorrhea at puberty, sexual infantilism
- Short stature possible
- Streak gonads (bilateral, risk of malignancy)
Gonadal Cancer Risk: 25-30% - highest of all DSD - gonadoblastoma, dysgerminoma. The Y chromosome carries the TSPY gene (Testis Specific Protein Y) which is oncogenic in a dysgenetic gonad.
Investigations:
- Karyotype: 46,XY
- Serum testosterone: very low
- LH, FSH: markedly elevated (hypergonadotropic hypogonadism)
- AMH: undetectable
- Pelvic ultrasound/MRI: streak gonads, uterus present
- SRY and other gene mutation analysis
Management:
- Prophylactic bilateral gonadectomy as soon as diagnosed (high malignancy risk)
- Estrogen replacement at puberty (for feminization, bone density, cardiovascular health)
- Cyclic progesterone after estrogen to induce withdrawal bleeding if desired
- Fertility: possible via donor egg + own uterus (IVF/embryo transfer)
6.5 XY Partial (Mixed) Gonadal Dysgenesis
Karyotype: Usually 45,X/46,XY mosaicism (or structural Y abnormality)
Clinical Features:
- Highly variable phenotype (female, male, or ambiguous)
- Typically: one streak gonad + one dysgenetic testis
- Asymmetric genital development
- Short stature, possible Turner-like stigmata
- Risk of gonadoblastoma (intermediate, ~15%)
Management: Sex assignment based on functional potential; remove dysgenetic gonads; hormonal therapy as needed
6.6 Persistent Müllerian Duct Syndrome (PMDS)
Definition: 46,XY males with normal male external genitalia who have persistent Müllerian structures (uterus, fallopian tubes) due to deficiency of AMH or AMH receptor.
Pathophysiology:
- Mutations in AMH gene (chromosome 19p13) or AMH receptor (AMHR2) gene
- Testosterone synthesis and action are normal → male external genitalia
- AMH absent or non-functional → Müllerian structures fail to regress
Clinical Features:
- Normal male external genitalia
- Cryptorchidism (testes drawn into hernia sac or pelvis by Müllerian remnants)
- Discovered incidentally during hernia repair or orchiopexy when uterus/tubes are found
- Fertility may be impaired due to cryptorchidism and associated traction on vas deferens
Investigations:
- AMH level: very low or undetectable (AMH gene mutation)
- AMH receptor gene mutation analysis
- Imaging: uterus/tubes on MRI
Management:
- Orchiopexy (preserving vas deferens, which may be adherent to Müllerian structures)
- Müllerian remnant removal where technically feasible without vas injury
7. SEX CHROMOSOME DSD
7.1 Turner Syndrome (45,X)
Karyotype: 45,X (monosomy X); or variants (45,X/46,XX mosaicism, ring X, isochromosome Xq, deletions)
Pathophysiology:
- Absence of second sex chromosome → bilateral streak gonads (no follicles; cortical stroma only)
- Streak gonads produce no estrogen or AMH → Müllerian structures present (uterus present but infantile), no feminization at puberty
- Haploinsufficiency of genes on X (particularly SHOX) → short stature, skeletal anomalies
Clinical Features:
- Short stature (most consistent feature; average adult height ~147 cm)
- Webbed neck (pterygium colli)
- Low posterior hairline
- Shield chest, wide-spaced nipples
- Lymphedema (hands and feet) at birth
- Cubitus valgus (wide carrying angle)
- Short 4th metacarpals
- Coarctation of the aorta (15-20%), bicuspid aortic valve (30%)
- Gonadal dysgenesis → primary amenorrhea, sexual infantilism, infertility
- Renal anomalies (horseshoe kidney, duplicated collecting system)
- Hypothyroidism (30%), autoimmune
- Cognitive: generally normal IQ but impaired spatial reasoning
Investigations:
- Karyotype (peripheral blood, 25-30 cells)
- Pelvic ultrasound: streak gonads, infantile uterus
- LH, FSH: markedly elevated
- Estradiol: very low
- Echocardiogram (cardiac anomalies)
- Renal ultrasound
- Thyroid function tests
- Audiometry (sensorineural hearing loss)
- Bone age X-ray
Management:
- Growth hormone therapy: Started as early as possible (FDA approved); significantly improves final height (gains of 5-10 cm)
- Estrogen replacement: low-dose estrogen started at ~11-12 years (before expected puberty) to induce feminization; progesterone added after 1-2 years for cyclic withdrawal bleeding
- Cardiac surveillance: MRI aorta every 5-10 years (risk of aortic dissection)
- Fertility: very rarely spontaneous pregnancy in mosaic forms; egg donation/IVF for most; careful cardiac evaluation before pregnancy (aortic dissection risk)
- Hormone replacement maintained through menopause age
7.2 Klinefelter Syndrome (47,XXY)
Karyotype: 47,XXY (80%); variants 48,XXXY, 49,XXXXY, mosaics (46,XY/47,XXY)
Pathophysiology:
- Additional X chromosome → progressive hyalinization and fibrosis of seminiferous tubules post-puberty → azoospermia
- Leydig cell dysfunction → testosterone deficiency (variable; many have low-normal T)
- Elevated LH, FSH (primary hypogonadism)
Clinical Features:
- Tall stature (long legs), eunuchoid habitus
- Small, firm testes (< 4 mL in adults; fibrosis)
- Azoospermia - infertility in virtually all
- Gynecomastia (due to ↑ E2:T ratio)
- Reduced facial and body hair
- Sparse pubic hair
- Often normal or near-normal male phenotype (many undiagnosed)
- Learning disabilities (reading/language difficulties)
- Increased risk of breast cancer, autoimmune disease, osteoporosis
Investigations:
- Karyotype: 47,XXY
- Testosterone: low to low-normal
- LH, FSH: markedly elevated
- Estradiol: elevated
- Semen analysis: azoospermia
- Testicular biopsy: hyalinized tubules, absence of spermatogenesis in most areas
Management:
- Testosterone replacement therapy: IM testosterone enanthate/cypionate 200 mg every 2-3 weeks, or transdermal formulations; normalize libido, energy, bone density, reduce gynecomastia
- Fertility: Microdissection testicular sperm extraction (microTESE) can retrieve sperm in 30-70% of cases for use with ICSI; best outcomes before age 35
- Gynecomastia: subcutaneous mastectomy if significant
- Psychological support; occupational therapy for learning disabilities
8. MANAGEMENT PRINCIPLES FOR DSD - MULTIDISCIPLINARY APPROACH
Modern management of DSD requires a dedicated multidisciplinary team (MDT):
- Pediatric endocrinologist
- Pediatric urologist / pediatric surgeon
- Clinical psychologist / psychiatrist
- Geneticist
- Radiologist
- Neonatologist
- Social worker
- Ethicist (where needed)
- Adult endocrinologist (for transition)
Key Ethical and Management Principles (Post-2006 Consensus):
- Avoid irreversible surgical procedures in infancy/childhood unless medically necessary - allow the individual to participate in gender decision
- Openness and full disclosure to the family (and eventually to the patient) about the diagnosis - the older practice of withholding the karyotype is no longer acceptable
- Gender assignment should be based on: karyotype, gonadal histology, degree of virilization, potential for adult sexual function, fertility potential, risk of malignancy, and family preference
- Gonadectomy timing: when indicated, balance malignancy risk vs. endocrine function preservation
- Psychosocial support: ongoing, from diagnosis through adulthood; address gender identity, sexual function, and disclosure
- Fertility counseling: early discussion even if fertility is not immediately possible
- Long-term follow-up: transition from pediatric to adult care is critical
Malignancy Risk Summary by Condition:
| Condition | Gonadal Cancer Risk | Timing of Gonadectomy |
|---|---|---|
| Complete AIS (CAIS) | ~10% (post-pubertal) | After puberty (to allow breast development) |
| PAIS | Variable (~15-50% for intra-abdominal gonads) | Individualized |
| Complete gonadal dysgenesis (Swyer) | 25-30% | Immediately at diagnosis |
| Partial/mixed gonadal dysgenesis | ~15% | Individualized |
| Turner syndrome (streak gonads) | ~1-2% (gonadoblastoma) | Low risk; no routine gonadectomy unless Y material present |
| Ovotesticular DSD | ~4% | Remove discordant gonadal tissue |
9. SUMMARY TABLE: KEY DISTINGUISHING FEATURES
| Condition | Karyotype | Gonads | Müllerian Structures | External Genitalia | Key Diagnostic Marker |
|---|---|---|---|---|---|
| CAH (21-OH def.) | 46,XX | Ovaries | Present | Virilized (Prader I-V) | ↑17-OHP, salt wasting |
| CAH (11β-OH def.) | 46,XX | Ovaries | Present | Virilized | ↑11-deoxycortisol, HTN |
| Complete AIS | 46,XY | Testes (intra-abd.) | Absent | Female | ↑T, 46,XY in female, AR mutation |
| Partial AIS | 46,XY | Testes | Absent | Ambiguous | ↑T, AR mutation |
| 5α-Reductase def. | 46,XY | Testes (undescended) | Absent | Ambiguous (hypospadias) | T:DHT ratio >30:1 |
| Swyer Syndrome | 46,XY | Streak gonads | Present | Female | ↑FSH/LH, undetectable AMH |
| 17β-HSD def. | 46,XY | Testes | Absent | Female/ambiguous | ↑androstenedione:T ratio |
| 17α-OH def. | 46,XY or XX | Testes/Ovaries | Absent (XY)/Present (XX) | Female (both) | HTN, ↓cortisol, ↑DOC |
| PMDS | 46,XY | Testes | Present (paradox) | Male | Undetectable AMH |
| Turner Syndrome | 45,X | Streak gonads | Present (infantile) | Female | ↑FSH/LH, short stature, stigmata |
| Klinefelter Syndrome | 47,XXY | Small firm testes | Absent | Male | Azoospermia, ↑FSH/LH, ↓T |
| Ovotesticular DSD | Variable | Ovotestis or mixed | Variable | Variable/ambiguous | Gonad biopsy |
Sources:
- Smith & Tanagho's General Urology, 19th Edition - Chapter 43: Disorders of Sex Development
- Campbell-Walsh-Wein Urology - Chapter on Intersex/DSD and Chapter on CAH
- Harrison's Principles of Internal Medicine 22E (2025) - Chapters on Gonadal Disorders and Androgen Insensitivity
- Creasy & Resnik's Maternal-Fetal Medicine - Revised Classification for DSD
This is a shared conversation. Sign in to Orris to start your own chat.