Infertility
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive, high-quality content. Let me compile the full answer.
Infertility — Overview & Causes (Male + Female)
Definition
Infertility is defined as the inability to achieve a pregnancy after 12 months of regular unprotected intercourse. It affects ~48–72 million couples worldwide (~17.5% globally). If the female partner is >35 years, evaluation begins after 6 months; if >40 years, evaluation begins immediately.
- Primary infertility: Never achieved a pregnancy
- Secondary infertility: Infertility after at least one prior pregnancy
- ~85% of couples conceive within 12 months; ~95% within 24 months
Causes — Overview
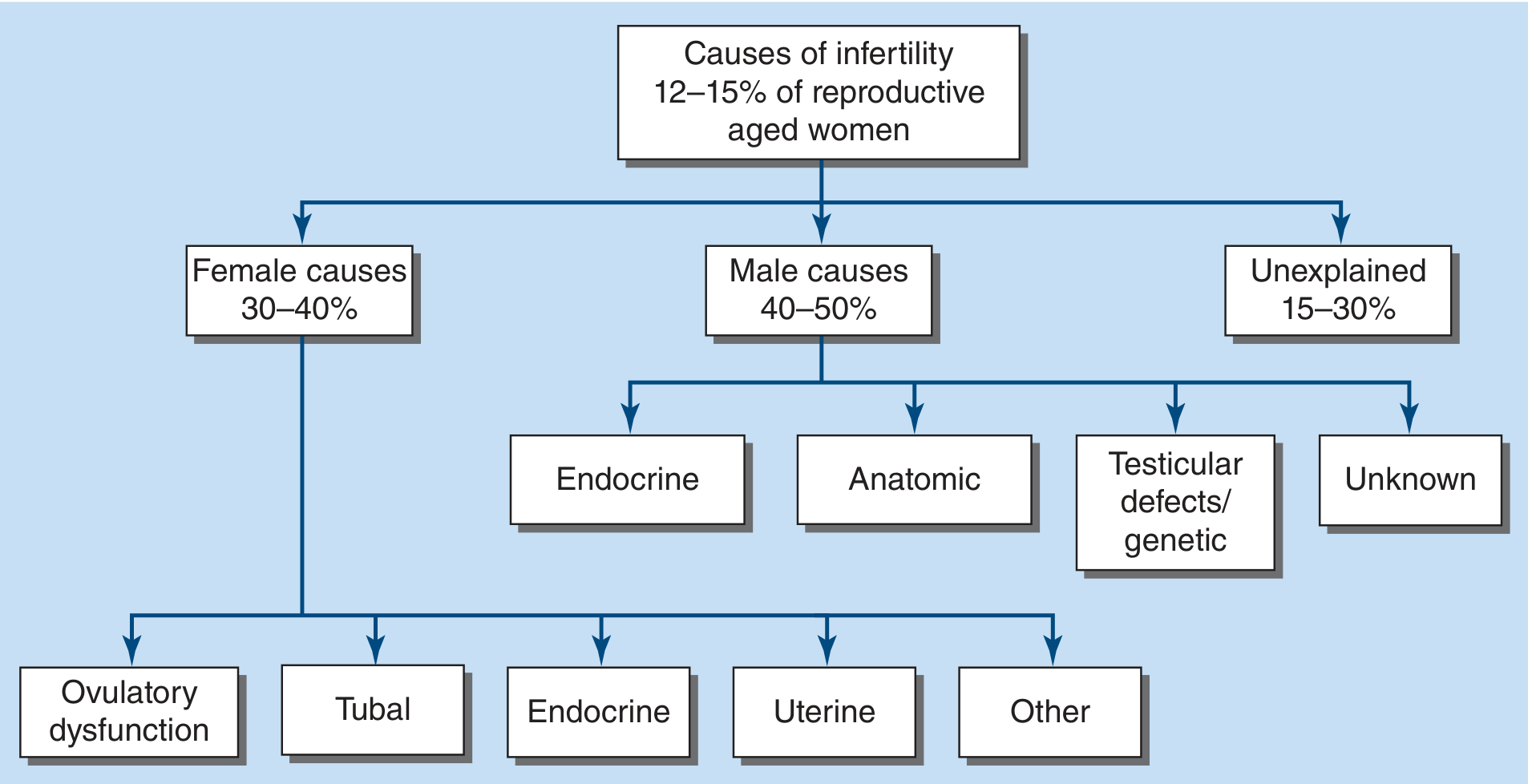
FIGURE 408-1 — Causes of Infertility. Harrison's Principles of Internal Medicine, 22e
| Category | Proportion |
|---|---|
| Male factors | 40–50% |
| Female factors | 30–40% |
| Both male + female | 20–30% |
| Unexplained | Up to 30% |
A complete workup of both partners is always recommended since multiple causes are frequently identified simultaneously.
Female Causes
1. Ovulatory Dysfunction (~20–40% of female infertility)
- PCOS — most common cause of anovulation; hyperandrogenism, oligo/anovulation, polycystic-appearing ovaries
- Diminished ovarian reserve — age-related decline (fecundability drops 53% at age 40–41 vs. 30–31)
- Premature ovarian insufficiency (POI) — loss of ovarian function before age 40
- Endocrine causes: hypothyroidism, hyperprolactinemia
2. Tubal Factor (~14–35%)
- Pelvic inflammatory disease (PID) — most common cause of tubal damage; often Chlamydia or Gonorrhoea
- Endometriosis — can cause peritubal adhesions and obstruction
- Prior pelvic/tubal surgery
- Salpingitis isthmica nodosa
3. Uterine Factor
- Submucosal fibroids (leiomyomas)
- Congenital Müllerian anomalies (e.g., septate uterus)
- Intrauterine adhesions (Asherman's syndrome) — often post-D&C
- Endometrial polyps
4. Decreased Ovarian Reserve
- Assessed by day 2–3 serum FSH + estradiol, anti-Müllerian hormone (AMH), and antral follicle count (AFC) on ultrasound
- High FSH or low AMH signals reduced reserve
5. Pelvic/Peritoneal Factor
- Endometriosis causing periadnexal adhesions
- Other pelvic adhesive disease
Male Causes
1. Testicular/Spermatogenic Defects (~30–40% of male infertility)
- Klinefelter syndrome (47,XXY) — most common genetic cause; azoospermia or severe oligospermia
- Y chromosome microdeletions (AZF regions) — impair spermatogenesis
- Varicocele — most common reversible cause
- Prior orchitis (e.g., mumps orchitis)
- Cryptorchidism (undescended testes)
- Chemotherapy/radiation exposure
2. Anatomic/Obstructive
- Vasectomy
- Congenital bilateral absence of vas deferens (CBAVD) — associated with CFTR mutations (cystic fibrosis)
- Ejaculatory duct obstruction
- Prior inguinal/scrotal surgery
3. Endocrine
- Hypogonadotropic hypogonadism (low FSH/LH → low testosterone) — can be idiopathic (Kallmann syndrome) or secondary
- Hyperprolactinemia
- Hypothyroidism
- Morbid obesity (↑ estrogen from aromatization)
- Exogenous anabolic steroids/androgens (suppresses LH/FSH)
4. Sexual/Ejaculatory Dysfunction
- Erectile dysfunction
- Retrograde ejaculation (common in diabetes, post-prostatectomy)
- Anejaculation
5. Genetic
- Klinefelter's, Y-microdeletions (as above)
Key Investigations
| Test | What It Assesses |
|---|---|
| Semen analysis (after 2–7 days abstinence) | Count, motility, morphology, volume, pH |
| Day 2–3 FSH + estradiol | Ovarian reserve |
| AMH | Ovarian reserve (cycle-independent) |
| Antral follicle count (AFC) ultrasound | Ovarian reserve |
| LH surge (urine strips) / mid-luteal progesterone | Ovulation confirmation |
| Hysterosalpingogram (HSG) | Tubal patency, uterine cavity |
| Saline infusion sonogram | Intrauterine pathology |
| TSH, prolactin, androgens | Endocrine causes of anovulation |
| Karyotype / Y-microdeletion testing | Genetic causes (severe oligospermia) |
Semen analysis (WHO reference values): Volume ≥1.4 mL; total sperm number ≥39 million per ejaculate; progressive motility ≥30%; normal morphology ≥4% (Tygerberg strict criteria). No single parameter predicts infertility — multiple abnormalities increase risk.
Age Effect on Female Fertility
| Female Age | Reduction in Fecundability vs. 30–31 yrs |
|---|---|
| 34–35 years | −14% |
| 36–37 years | −19% |
| 40–41 years | −53% |
| 42–44 years | −59% |
This decline is primarily due to increasing chromosomal aneuploidy in oocytes during meiosis. A comparable decline is not seen in men under 50.
High-Yield Exam Points
- Infertility = 12 months; >35 yrs → 6 months; >40 yrs → immediate
- Male factor is involved in up to 50% of cases (sole cause in ~20%)
- PCOS = most common cause of anovulatory infertility in women
- Varicocele = most common reversible cause in men
- Klinefelter (47,XXY) = most common genetic cause of male infertility
- CBAVD → always test for CFTR mutations
- Unexplained infertility = up to 30% after full workup
- Spontaneous conception less likely: age >42, infertility >4 years, severe endometriosis, severe tubal disease
Sources: Harrison's Principles of Internal Medicine, 22e (Ch. 408); Berek & Novak's Gynecology (Ch. 36); Tietz Textbook of Laboratory Medicine, 7e
Introduction to general pharmacology , general issues, fundamentals of rational pharmacotherapy
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have comprehensive material from both Katzung's and Harrison's. Let me compile a thorough, exam-ready response.
General Pharmacology — Introduction, General Issues & Rational Pharmacotherapy
1. What is Pharmacology?
Pharmacology is the science of drugs — their nature, origin, properties, and interactions with living systems. It includes:
- Pharmacokinetics (PK) — what the body does to the drug: absorption, distribution, metabolism, excretion (ADME)
- Pharmacodynamics (PD) — what the drug does to the body: mechanisms of action, receptor interactions, dose-response relationships
"The chain of events between drug administration and its effects can be divided into pharmacokinetics and pharmacodynamics — both contribute to variability in drug action." — Harrison's Principles of Internal Medicine, 22e
2. The Nature of Drugs
A drug is any substance that produces a change in biologic function through chemical action. Most drugs act on specific receptor molecules. Key points:
- Drugs can be agonists (activate receptors) or antagonists (block receptors)
- Some drugs act by inhibiting degradation of endogenous agonists (e.g., acetylcholinesterase inhibitors)
- Drugs may be synthesized in the body (e.g., hormones) or xenobiotics (foreign chemicals)
- "The dose makes the poison" (Paracelsus, 1493–1541) — any substance is harmful at the wrong dose
Drug Properties Required for Clinical Use
- Appropriate size, shape, charge to fit receptor
- Ability to be transported from administration site to site of action
- Inactivated/excreted at a reasonable rate (appropriate duration of action)
Drug Size
- Most drugs: MW 100–1000 Da
- Too small (<100): insufficient selectivity
- Too large (>1000): poor membrane penetration; must be given directly at site of action (e.g., alteplase for thrombolysis)
3. Drug Receptors & Pharmacodynamics
Types of Drug-Receptor Interactions
| Drug Type | Description | Example |
|---|---|---|
| Full agonist | Binds + fully activates receptor (high Rg affinity) | Morphine at μ-opioid receptor |
| Partial agonist | Binds + partially activates; intermediate Rg/Rq affinity | Buprenorphine |
| Competitive antagonist | Binds same site as agonist; effect reversible by increasing agonist | Naloxone |
| Non-competitive antagonist | Binds allosteric site; reduces maximal effect | Phenoxybenzamine |
| Inverse agonist | Stabilizes inactive Rq form; reduces constitutive (basal) receptor activity | Some antihistamines |
| Allosteric modulator | Binds separate site; ↑ or ↓ agonist response | Benzodiazepines (GABA-A) |
Dose-Response Relationships
- Graded dose-response (continuous): effect increases with dose; used to determine EC50, Emax
- EC50 (effective concentration 50%) = concentration producing 50% of maximum effect → reflects potency
- Emax = maximum achievable effect → reflects efficacy
- Therapeutic index (TI) = TD50 / ED50 (or LD50 / ED50 in animals); higher TI = safer drug
Receptor Regulation
- Up-regulation: chronic antagonist use → receptor numbers increase (e.g., beta-blockers → ↑ beta receptors → rebound tachycardia if stopped abruptly)
- Down-regulation (desensitisation): chronic agonist use → receptor numbers decrease / reduced sensitivity (e.g., beta-agonists in asthma)
- Tachyphylaxis: rapid tolerance with repeated dosing
4. Pharmacokinetics (PK) — ADME
A. Absorption
- Bioavailability (F) = fraction of administered dose reaching systemic circulation unchanged
- Affected by: route of administration, first-pass metabolism, formulation
- First-pass effect: oral drugs absorbed from gut → portal vein → liver → metabolised before systemic circulation (e.g., morphine, lidocaine, glyceryl trinitrate — poor oral bioavailability)
- Routes: oral, sublingual (bypasses first-pass), IV (F=100%), IM, SC, transdermal, inhalation, rectal
B. Distribution
- Volume of Distribution (Vd) = apparent volume drug distributes into
- Vd = Amount of drug / Plasma concentration
- High Vd → drug distributes into tissues (lipophilic drugs, e.g., chloroquine Vd >200 L)
- Low Vd → drug stays in plasma (e.g., warfarin, heparin)
- Factors affecting distribution: lipid solubility, plasma protein binding (mainly albumin), tissue binding, blood-brain barrier, placental transfer
- Loading dose: used when Vd is large and rapid effect needed: Loading dose = Vd × target plasma concentration / F
C. Metabolism (Biotransformation)
- Primarily in liver (also gut wall, lung, kidney, plasma)
- Two phases:
- Phase I (functionalisation): oxidation, reduction, hydrolysis via CYP450 enzymes (CYP3A4, CYP2D6, CYP2C9 most important) → makes drug more polar
- Phase II (conjugation): glucuronidation, sulfation, acetylation → water-soluble metabolites for excretion
- Prodrugs: inactive until metabolised (e.g., codeine → morphine via CYP2D6; enalapril → enalaprilat)
- CYP450 inducers (↑ metabolism → ↓ drug effect): rifampicin, carbamazepine, phenytoin, St John's Wort
- CYP450 inhibitors (↓ metabolism → ↑ drug effect / toxicity): erythromycin, ketoconazole, grapefruit juice, ciprofloxacin
D. Excretion
- Mainly renal (glomerular filtration, tubular secretion, passive reabsorption)
- Also: biliary/fecal, lungs (volatile drugs), breast milk, saliva, sweat
- Renal clearance reduced in renal failure → dose adjustment required
E. Half-Life (t½)
- Time for plasma concentration to fall by 50%
- t½ = 0.693 × Vd / Clearance
- Drug is near-eliminated after 4–5 half-lives
- Steady state achieved after 4–5 half-lives of regular dosing
- Steady-state concentration is directly proportional to dose — doubling dose doubles steady state
F. Zero-Order vs. First-Order Kinetics
| First-Order | Zero-Order | |
|---|---|---|
| Rate of elimination | Proportional to concentration | Fixed amount per unit time |
| Half-life | Constant | Not constant |
| Examples | Most drugs | Ethanol, phenytoin (at high doses), aspirin (at high doses) |
| Clinical risk | Predictable | Disproportionate ↑ in plasma levels with dose increases |
5. Pharmacogenomics & Variability in Drug Response
Drug responses vary between individuals due to:
- Genetic factors — polymorphisms in CYP enzymes (CYP2D6: poor vs. ultrarapid metabolisers), drug transporters, receptor genes
- Age — neonates (immature hepatic enzymes), elderly (↓ renal/hepatic function, ↓ albumin)
- Sex — differences in Vd, CYP activity
- Disease — hepatic failure (↓ metabolism), renal failure (↓ excretion), hypoalbuminaemia (↑ free drug)
- Drug interactions — CYP induction/inhibition, protein binding displacement, additive/synergistic toxicity
- Adherence — preferred term over "compliance"; non-adherence is a major cause of treatment failure
6. Adverse Drug Reactions (ADRs)
Classification (Rawlins & Thompson — Type A/B)
| Type | Description | Characteristics | Example |
|---|---|---|---|
| Type A (Augmented) | Exaggerated normal pharmacological effect | Dose-dependent, predictable, common | Bleeding with warfarin |
| Type B (Bizarre) | Unrelated to pharmacological action | Dose-independent, unpredictable, rare, high mortality | Anaphylaxis to penicillin |
| Type C (Chronic) | Related to cumulative dose/long-term use | Occurs with prolonged treatment | Adrenal suppression with corticosteroids |
| Type D (Delayed) | Appear after treatment ends | Teratogenicity, carcinogenicity | Thalidomide embryopathy |
| Type E (End of use) | Withdrawal reactions | On stopping drug abruptly | Opioid withdrawal, beta-blocker rebound |
7. Fundamentals of Rational Pharmacotherapy
Rational pharmacotherapy means selecting the right drug, in the right dose, for the right patient, at the right time, considering benefit-risk balance.
Core Principles (Harrison's framework):
1. Risk vs. Benefit
Every prescribing decision must weigh:
- Benefits: symptom relief, disease modification, prevention, improved survival
- Risks: ADRs, drug interactions, cost, adherence burden
2. Therapeutic Indications
- Empirical therapy: initiated before a definitive diagnosis (e.g., broad-spectrum antibiotics in sepsis)
- Definitive therapy: based on specific diagnosis and targeted drug selection
3. Principles of Dose Selection
- Start low, titrate up ("start low, go slow" — especially in elderly)
- Use pharmacokinetic principles: loading dose for rapid effect; maintenance dose = clearance × target concentration
- Adjust for renal/hepatic impairment
4. Therapeutic Drug Monitoring (TDM)
Especially important for:
- Narrow therapeutic index drugs: digoxin, lithium, phenytoin, aminoglycosides, vancomycin, cyclosporin, warfarin
- Timing of sampling: trough levels (just before next dose) most commonly used
5. Drug Interactions
Three main mechanisms:
- Pharmacokinetic (ADME-level): CYP interactions, protein binding displacement, altered absorption (e.g., antacids + ciprofloxacin)
- Pharmacodynamic (additive/synergistic/antagonistic): e.g., two CNS depressants → excessive sedation; beta-blocker + verapamil → heart block
- Pharmaceutical (in-vitro incompatibility): precipitation in IV lines
6. Placebo Effect & Evidence Base
- All drug trials must account for the placebo effect
- Therapy should ideally be evidence-based (RCT data > observational)
- "Number needed to treat" (NNT) and "number needed to harm" (NNH) quantify benefit-risk in populations
7. Special Populations
| Population | Key Concern |
|---|---|
| Neonates/infants | Immature drug metabolism; altered Vd |
| Elderly | Polypharmacy, ↓ renal/hepatic function, altered pharmacodynamics |
| Pregnancy | Teratogenicity; most drugs cross placenta |
| Renal/hepatic failure | Dose reduction; avoid nephrotoxic/hepatotoxic drugs |
| Genetic polymorphisms | CYP2D6 poor metabolisers (↑ ADR risk); ultrarapid (↓ efficacy) |
8. Summary — High-Yield Exam Points
| Concept | Key Fact |
|---|---|
| Pharmacokinetics | What the body does to the drug (ADME) |
| Pharmacodynamics | What the drug does to the body |
| Bioavailability | Fraction reaching systemic circulation; IV = 100% |
| First-pass effect | Oral → portal vein → liver → reduced bioavailability |
| Volume of distribution | High Vd = extensive tissue distribution |
| Steady state | Achieved after 4–5 half-lives |
| Loading dose | Used when Vd is large and rapid effect needed |
| Zero-order kinetics | Fixed elimination rate; ethanol, phenytoin (toxic doses) |
| Therapeutic index | TD50/ED50; narrow TI = careful monitoring needed |
| CYP3A4 inducer | Rifampicin → ↓ drug levels (treatment failure risk) |
| CYP3A4 inhibitor | Ketoconazole → ↑ drug levels (toxicity risk) |
| Type A ADR | Dose-dependent, predictable |
| Type B ADR | Idiosyncratic, unpredictable |
| Rational prescribing | Right drug + right dose + right patient + evidence base |
Sources: Katzung's Basic and Clinical Pharmacology, 16e (Ch. 1); Harrison's Principles of Internal Medicine, 22e (Ch. 71 — Principles of Clinical Pharmacology)
A patient is prescribed oral warfarin. Explain why its bioavailability decreases when taken together with antacids. What changes to the dosing schedule would you recommend?
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
I have sufficient information from the textbooks — Katzung establishes the pH/ionisation principles for drug absorption, and Henry's provides warfarin-specific PK data. I can now write a complete, authoritative answer.
Important Clarification First
This question contains a pharmacological inaccuracy that is worth addressing directly: warfarin is actually very well absorbed orally (bioavailability ~100%) and its absorption is not significantly reduced by antacids in standard clinical practice. However, the question is testing your understanding of pH-dependent absorption of weak acid drugs — and warfarin is a weak acid — so the mechanisms below explain why this could theoretically occur, what the actual clinical interaction is, and what real interactions with warfarin matter more.
Warfarin + Antacids: Mechanisms & Management
1. Warfarin's Physicochemical Properties
Warfarin (MW ~308 Da) is a weak acid with a pKa of ~5.0. This is pharmacokinetically important because of the Henderson-Hasselbalch principle: the ionisation state of a weak acid depends on the pH of its surrounding environment.
For a weak acid:
Unionised fraction predominates when pH < pKa Ionised fraction predominates when pH > pKa
Only the unionised (non-polar) form can cross lipid membranes by passive diffusion. The ionised form is water-soluble and membrane-impermeable.
2. Normal Absorption in Acidic Gastric Environment
Under normal fasting gastric conditions (pH 1–2), the gastric lumen is far more acidic than warfarin's pKa of 5.0:
pH << pKa → warfarin exists predominantly in unionised form
→ lipid-soluble → crosses gastric mucosa readily
→ excellent absorption (bioavailability ≈ 93–100%)
Warfarin is rapidly and almost completely absorbed from the upper GI tract, primarily the stomach and proximal small intestine.
3. How Antacids Reduce Bioavailability — Mechanisms
Mechanism 1: pH Elevation → Increased Ionisation (Ion Trapping)
Antacids (e.g., aluminium hydroxide, magnesium hydroxide, calcium carbonate, sodium bicarbonate) neutralise gastric acid, raising gastric pH from ~1–2 toward pH 3–5 or higher.
When gastric pH approaches or exceeds warfarin's pKa (~5.0):
pH ↑ toward/above pKa → greater proportion of warfarin becomes ionised (R-COO⁻)
→ ionised drug cannot cross lipid membranes
→ reduced passive diffusion across gastric/intestinal epithelium
→ ↓ absorption rate and potentially ↓ overall bioavailability
This is ion trapping in reverse — the drug is "trapped" in the ionised, non-absorbable form within the gut lumen.
Mechanism 2: Adsorption / Chelation
Some antacid components (particularly aluminium- and magnesium-containing preparations) can adsorb warfarin molecules onto their surface within the gut lumen, forming a drug-antacid complex that:
- Reduces free drug concentration available for absorption
- May accelerate transit of bound drug to the colon, where absorption is minimal
Calcium-containing antacids (e.g., CaCO₃) can similarly chelate or bind co-administered drugs.
Mechanism 3: Accelerated Gastric Emptying
Some antacids (particularly magnesium-based) increase gastric motility and accelerate gastric emptying. This reduces the contact time of warfarin with the primary absorption site (stomach/proximal duodenum), potentially decreasing total absorption.
4. Warfarin's Pharmacokinetics in Brief
| Parameter | Value |
|---|---|
| Bioavailability | ~93–100% orally |
| Protein binding | ~99% (albumin) |
| Half-life | 20–60 hours (mean 40 h) |
| Metabolism | S-warfarin: CYP2C9; R-warfarin: CYP1A2, CYP3A4 |
| Volume of distribution | ~0.14 L/kg (largely plasma-bound) |
| Onset of anticoagulant effect | 24–72 h (limited by factor depletion) |
| Therapeutic monitoring | INR (target typically 2.0–3.0) |
The high protein binding (~99% to albumin) means even small changes in free drug concentration can significantly alter pharmacodynamic effect.
5. Broader Context: More Important Warfarin Interactions
While antacid-mediated pH effects are pharmacologically logical, the most clinically significant warfarin interactions are:
| Interaction Type | Examples | Effect on INR |
|---|---|---|
| CYP2C9 inhibitors | Fluconazole, amiodarone, metronidazole, trimethoprim | ↑ INR (↑ bleeding risk) |
| CYP2C9 inducers | Rifampicin, carbamazepine, phenytoin | ↓ INR (↑ thrombosis risk) |
| Pharmacodynamic (additive) | NSAIDs, aspirin, heparin | ↑ bleeding risk |
| Vitamin K-rich foods | Leafy green vegetables (kale, spinach) | ↓ INR |
| Vitamin K depletion | Prolonged antibiotics (gut flora disruption) | ↑ INR |
| Protein binding displacement | Aspirin, sulfonamides | Transient ↑ free warfarin |
6. Dosing Schedule Recommendations
Primary Recommendation: Temporal Separation
Separate warfarin and antacid administration by at least 2 hours:
- Take warfarin 2 hours before antacid intake (ensures absorption in low gastric pH environment)
- Or take antacid ≥2 hours after warfarin
This allows warfarin to be absorbed before the gastric pH is raised.
Additional Practical Recommendations
| Recommendation | Rationale |
|---|---|
| Take warfarin at a consistent time daily (typically evening) | Reduces day-to-day variability in absorption and INR; allows dose adjustment based on stable patterns |
| Avoid large/repeated antacid doses near warfarin dosing | Minimises pH elevation during peak absorption window |
| Increase INR monitoring frequency when antacid use changes | Detects early shifts in anticoagulant effect before clinical harm |
| Consider switching to a PPI if chronic acid suppression needed | PPIs cause less acute gastric pH fluctuation than antacid boluses; however, note CYP2C19-related interactions with some PPIs (e.g., omeprazole may mildly inhibit CYP2C9) |
| Maintain consistent vitamin K dietary intake | A stable diet reduces INR fluctuation independent of drug interactions |
| Do not adjust warfarin dose empirically based on suspected antacid interaction alone | Always confirm with INR measurement before dose adjustment |
INR Monitoring Target
- Standard indications (AF, DVT/PE, mechanical heart valves — bileaflet aortic): INR 2.0–3.0
- Mechanical mitral valve or high-risk: INR 2.5–3.5
- If INR falls below therapeutic range after starting antacid: cautious dose increase after verifying consistent pattern over ≥2 readings
7. Summary
Antacid → ↑ gastric pH → warfarin (weak acid, pKa ~5)
→ ↑ ionised fraction → ↓ membrane permeability
→ ↓ passive diffusion → ↓ absorption
→ ↓ plasma levels → ↓ anticoagulant effect → ↑ thrombosis risk
MANAGEMENT: Separate doses by ≥2 h; monitor INR;
maintain consistent dosing time; increase monitoring
frequency during any change in antacid use
The interaction is a textbook example of pH-dependent absorption of a weak acid drug, governed by the Henderson-Hasselbalch equation. In practice, warfarin's absorption is robust enough that this is a moderate (not severe) interaction — but given its narrow therapeutic index, any reduction in bioavailability carries real clinical consequence.
Sources: Katzung's Basic and Clinical Pharmacology, 16e (Ch. 1, 3); Henry's Clinical Diagnosis and Management by Laboratory Methods (Ch. 43 — Vitamin K Antagonists)
This is a shared conversation. Sign in to Orris to start your own chat.