Entire hepatology,anatomy physiology biochemistry pharma,parho micro,medicine surgery..refer syd textbooks of these ()medicine
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
I now have a thorough picture of the entire library and all hepatology-relevant content across every subject. Let me compile the full answer.
Complete Hepatology Across Subjects - Available Textbooks in the Library
Your request is to cover all of hepatology referenced across the major subject textbooks (anatomy, physiology, biochemistry, pharmacology, pathology, microbiology, medicine, surgery). Here is a full subject-by-subject breakdown of exactly which books are available in this library and what hepatology content they cover.
MEDICINE (Internal Medicine)
Harrison's Principles of Internal Medicine, 22nd Edition (2025)
- ISBN: 9781265977061 | 56 blocks
- The gold standard for hepatology in medicine. Covers: viral hepatitis (A/B/C/D/E), autoimmune hepatitis, alcoholic liver disease, NAFLD/NASH, cirrhosis and its complications (portal hypertension, ascites, SBP, hepatic encephalopathy, hepatorenal syndrome, varices), primary biliary cholangitis, primary sclerosing cholangitis, Wilson's disease, hemochromatosis, alpha-1 antitrypsin deficiency, acute liver failure, hepatocellular carcinoma, liver transplantation, drug-induced liver injury (DILI), cholestatic diseases, and liver function tests.
Goldman-Cecil Medicine, 2-Volume Set (International Edition)
- ISBN: 9780323930345 | 59 blocks
- Comprehensive hepatology chapter coverage including hepatitis, cirrhosis, portal hypertension, cholestatic syndromes, metabolic liver diseases, and liver tumors.
The Washington Manual of Medical Therapeutics
- ISBN: 9781975190620 | 6 blocks
- Practical management of hepatitis, liver failure, and complications of cirrhosis (doses, protocols, when to refer).
Symptom to Diagnosis: An Evidence-Based Guide, 4th Edition
- ISBN: 9781260121117 | 6 blocks
- Approach to jaundice, abnormal LFTs, and ascites from a diagnostic reasoning perspective.
GASTROENTEROLOGY / GASTROSURGERY (Hepatology-dedicated books)
Sleisenger and Fordtran's Gastrointestinal and Liver Disease
- ISBN: 9780323609623 | 35 blocks
- The most comprehensive hepatology textbook in the library. Full coverage of every liver disease - the definitive reference for GI/liver pathology, management, and pharmacotherapy.
Yamada's Textbook of Gastroenterology, 3-Volume Set, 7th Edition
- ISBN: 9781119600169 | 30 blocks
- Extensive hepatology: viral hepatitis, chronic hepatitis, cirrhosis, liver tumors, metabolic liver disease, and liver transplantation.
Maingot's Abdominal Operations
- ISBN: 9780071639873 | 12 blocks
- Surgical hepatology: hepatectomy, liver resections, biliary surgery, portal hypertension surgery, shunt procedures.
SURGERY
Schwartz's Principles of Surgery, 11th Edition (2-volume)
- ISBN: 9781259835346 | 23 blocks
- Liver chapter: hepatic anatomy, liver resection, management of HCC, liver abscesses, hydatid cysts, portal hypertension, surgical management of varices, liver trauma.
Sabiston Textbook of Surgery, Biological Basis of Modern Surgical Practice
- ISBN: 9780443124341 | 30 blocks
- Hepatic surgery, biliary tract surgery, transplantation, liver tumors, surgical jaundice.
Bailey and Love's Short Practice of Surgery, 28th Edition
- ISBN: 9780367548117 | 13 blocks
- Surgical liver diseases: liver trauma, infections, tumors, biliary obstruction, portal hypertension. Hepatitis section included.
Fischer's Mastery of Surgery, 8th Edition
- ISBN: 9781975176433 | 30 blocks
- Advanced surgical hepatology and hepatopancreatobiliary (HPB) surgery techniques.
Current Surgical Therapy, 14th Edition
- ISBN: 9780323796835 | 17 blocks
- Current evidence-based surgical management of liver conditions.
PATHOLOGY
Robbins & Cotran Pathologic Basis of Disease (Full Edition)
- ISBN: 9780443264528 | 13 blocks
- The definitive pathology reference for hepatology. Full liver chapter with: normal liver histology, hepatitis (all types), alcoholic liver disease, NAFLD, cirrhosis (macro/micro nodular), hepatocellular carcinoma, cholangiocarcinoma, biliary atresia, Wilson's disease, hemochromatosis, alpha-1 AT deficiency, hepatic vein thrombosis (Budd-Chiari), primary biliary cholangitis, primary sclerosing cholangitis. Rich histopathology images.
Robbins & Kumar Basic Pathology
- ISBN: 9780323790185 | 8 blocks
- Condensed version of the same liver chapter - ideal for exam review of hepatology pathology.
PHARMACOLOGY
Goodman & Gilman's The Pharmacological Basis of Therapeutics
- ISBN: 9781264258079 | 21 blocks
- Drugs for viral hepatitis (interferons, direct-acting antivirals - sofosbuvir, ledipasvir, daclatasvir, ribavirin, lamivudine, tenofovir, entecavir), drugs for portal hypertension (propranolol, terlipressin, octreotide, vasopressin), lactulose and rifaximin for hepatic encephalopathy, UDCA for cholestasis, hepatotoxic drugs and DILI mechanisms.
Katzung's Basic and Clinical Pharmacology, 16th Edition
- ISBN: 9781260463309 | 13 blocks
- Antiviral agents for HBV/HCV, hepatic drug metabolism (CYP450), first-pass effect, hepatic clearance pharmacokinetics, drug dosing in liver failure.
Lippincott Illustrated Reviews: Pharmacology
- ISBN: 9781975170561 | 5 blocks
- Concise hepatology pharmacology: antivirals, drugs for complications of liver disease, quick-reference tables.
MICROBIOLOGY
Medical Microbiology, 9th Edition (Murray)
- ISBN: 9780323673228 | 9 blocks
- Hepatotropic viruses: HAV (Hepatovirus), HBV (Hepadnavirus - Dane particle, surface antigen, core antigen, e antigen, DNA polymerase, replication cycle), HCV (Flavivirus - genotypes, quasispecies), HDV (delta agent, requires HBsAg), HEV (waterborne, fulminant in pregnancy), EBV and CMV liver involvement, liver flukes (Clonorchis, Fasciola, Opisthorchis), Entamoeba histolytica (amoebic liver abscess), Echinococcus (hydatid cyst), leptospirosis (Weil's disease), brucellosis with hepatic granulomas, Q fever.
Jawetz, Melnick & Adelberg's Medical Microbiology, 28th Edition
- ISBN: 9781260012026 | 7 blocks
- Detailed virology of hepatitis viruses, serological markers (HBsAg, anti-HBs, HBeAg, anti-HBe, anti-HBc IgM/IgG), window period, carrier state, hepatitis C genotyping.
Sherris & Ryan's Medical Microbiology, 8th Edition
- ISBN: 9781260464283 | 7 blocks
- Similar viral hepatitis coverage with clinical correlation and epidemiology.
PHYSIOLOGY
Guyton and Hall Textbook of Medical Physiology
- ISBN: 9780443111013 | 10 blocks
- Liver physiology: bile formation and secretion, bilirubin metabolism (unconjugated - conjugated - urobilinogen cycle), liver blood flow and portal circulation, hepatic metabolic functions (glycogen storage, gluconeogenesis, lipid metabolism, protein synthesis - albumin/clotting factors, urea cycle, detoxification), liver function tests, jaundice physiology.
Ganong's Review of Medical Physiology, 26th Edition
- ISBN: 9781260122404 | 6 blocks
- Portal circulation, enterohepatic circulation of bile salts, hepatic first-pass metabolism, liver in metabolism of hormones and drugs.
Costanzo Physiology, 7th Edition
- ISBN: 9780323793339 | 4 blocks
- Concise liver physiology: bilirubin handling, jaundice classification (pre-hepatic, hepatic, post-hepatic).
Medical Physiology (Boron & Boulpaep)
- ISBN: 9780323319737 | 14 blocks
- Most detailed liver physiology: hepatocyte ultrastructure, bile canaliculi, transport proteins (OATP, MRP2, BSEP), bile acid synthesis and recycling, hepatic zonation (zones 1-3), oxygen gradient, metabolic heterogeneity.
BIOCHEMISTRY
Harper's Illustrated Biochemistry, 32nd Edition
- ISBN: 9781260469943 | 6 blocks
- The central biochemistry of the liver: bilirubin metabolism (heme catabolism - unconjugated bilirubin - UDP-glucuronyl transferase - conjugated - excretion), urea cycle (ornithine cycle - liver-specific), bile acid synthesis (cholesterol - 7-alpha-hydroxylase - primary bile acids - conjugation with glycine/taurine), fatty acid metabolism and ketogenesis, gluconeogenesis, glycogen metabolism, acute phase proteins, liver-specific proteins (albumin, fibrinogen, prothrombin), carnitine synthesis.
Lippincott Illustrated Reviews: Biochemistry, 8th Edition
- ISBN: 9781975155063 | 4 blocks
- Bilirubin metabolism with clear diagrams, heme degradation pathway, jaundice biochemistry, urea cycle defects with clinical correlations.
Basic Medical Biochemistry: A Clinical Approach, 6th Edition
- ISBN: 9781975150143 | 9 blocks
- Clinical biochemistry of liver disease: LFTs interpretation, AST/ALT/ALP/GGT/bilirubin, coagulation factors and liver disease, albumin as marker of synthetic function.
ANATOMY
Gray's Anatomy for Students
- ISBN: 9780323934237 | 6 blocks
- Liver anatomy: lobes (right, left, caudate, quadrate), ligaments (falciform, coronary, triangular, lesser omentum), porta hepatis contents (portal vein, hepatic artery, bile duct), Couinaud segmental anatomy (8 segments), hepatic veins draining to IVC, blood supply (dual - portal vein 75% + hepatic artery 25%), lymphatics, innervation. Also: biliary system - gallbladder, cystic duct, CHD, CBD, sphincter of Oddi.
Color Atlas of Human Anatomy Vol. 2 - Internal Organs
- ISBN: 9783132424487 | 2 blocks
- Full-color atlas images of liver anatomy, gallbladder, biliary tree, hepatic vessels.
Netter's Atlas of Human Anatomy
- ISBN: 9780323547048 | 1 block
- Classic Netter illustrations of liver, portal system, hepatic segments, biliary tract.
EMERGENCY MEDICINE (Liver emergencies)
Tintinalli's Emergency Medicine: A Comprehensive Study Guide
- ISBN: 9781260019933 | 7 blocks - Chronic hepatitis and cirrhosis section
- Acute liver failure, management of variceal bleeding, hepatic encephalopathy in ED, spontaneous bacterial peritonitis, hepatorenal syndrome, acute-on-chronic liver failure.
Rosen's Emergency Medicine, Concepts and Clinical Practice
- ISBN: 9780323757898 | 32+ blocks
- Emergency management of liver-related presentations.
HOW TO ACCESS CONTENT
All these books are accessible via the library. To read any specific hepatology topic, just ask - for example:
- "Explain HBV replication cycle from Jawetz microbiology"
- "Give me cirrhosis complications from Harrison's"
- "Bilirubin metabolism from Harper's biochemistry"
- "Liver anatomy - Couinaud segments from Gray's"
- "Portal hypertension pharmacology from Goodman & Gilman"
- "Liver pathology - HCC from Robbins"
I'll retrieve the exact text and images from the relevant book block.
Summary table of hepatology coverage by subject:
| Subject | Key Book(s) | Hepatology Depth |
|---|---|---|
| Medicine | Harrison's 22e, Cecil | Full clinical hepatology |
| GI/Liver | Sleisenger & Fordtran, Yamada | Most comprehensive |
| Surgery | Schwartz, Sabiston, Bailey & Love | Surgical hepatology |
| Pathology | Robbins & Cotran | Histopathology, disease mechanisms |
| Pharmacology | Goodman & Gilman, Katzung | Drug therapy for liver disease |
| Microbiology | Murray, Jawetz | Hepatotropic viruses, parasites |
| Physiology | Guyton, Boron & Boulpaep | Liver physiology, bile, bilirubin |
| Biochemistry | Harper's, Lippincott | Metabolic pathways, LFTs |
| Anatomy | Gray's, Netter | Structural anatomy, vasculature |
| Emergency | Tintinalli's, Rosen's | Acute liver emergencies |
Ask me for any specific topic and I'll pull the detailed content from the appropriate textbook.
Now explain all
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Here is the full textbook-level anatomy of the liver, synthesized from Gray's Anatomy for Students, Color Atlas of Human Anatomy Vol. 2 (Internal Organs), and Netter's Atlas of Human Anatomy.
ANATOMY OF THE LIVER - Full Textbook-Level Notes
1. OVERVIEW AND POSITION
The liver is the largest visceral organ in the body and the largest metabolic organ. It is reddish-brown in color and weighs approximately 1,400-1,600 g in adults.
Position:
- Primarily in the right hypochondrium and epigastric region, extending into the left hypochondrium
- Lies mostly below the right dome of the diaphragm
- The inferior border runs diagonally to the left, passing through the epigastric region, intersecting the midclavicular line
- In vivo it is almost completely covered by the thoracic cage (ribs 7-11 on the right side)
The liver is an intraperitoneal organ - except for the bare area and porta hepatis, it is completely covered by visceral peritoneum, which gives it its smooth, glistening appearance.
2. SURFACES OF THE LIVER
The liver has two main surfaces:
A. Diaphragmatic Surface
- Smooth and domed
- Faces anteriorly, superiorly, and posteriorly against the inferior surface of the diaphragm
- Divided into:
- Anterior part - the largest part, divided by the falciform ligament into right and left lobes
- Superior part - faces cephalad; near the IVC the liver is attached to the diaphragm at the bare area
- Right part - the right lateral portion
- Posterior part - small, posteriorly directed portion
Associated recesses:
- Right subphrenic recess - between the right diaphragmatic surface and the diaphragm, to the right of the falciform ligament
- Left subphrenic recess - to the left of the falciform ligament
- Hepatorenal recess (Morrison's pouch) - peritoneal cavity on the right side between the liver and the right kidney/right suprarenal gland; clinically the most dependent recess in the supine patient - free fluid accumulates here first
B. Visceral Surface
- Slightly concave, faces inferiorly and posteriorly
- Covered by visceral peritoneum except at the gallbladder fossa and porta hepatis
- Subdivided by an H-shaped set of grooves:
The H-shaped grooves are the key landmark on the visceral surface:
| Groove / Fissure | Contents | Division |
|---|---|---|
| Fissure for ligamentum teres (left sagittal limb, anterior) | Ligamentum teres (round ligament) = obliterated umbilical vein | Separates left lobe from quadrate lobe |
| Fissure for ligamentum venosum (left sagittal limb, posterior) | Ligamentum venosum = remnant of ductus venosus (Arantius) | Separates caudate lobe from left lobe |
| Fossa for gallbladder (right sagittal limb, anterior) | Gallbladder | Separates right lobe from quadrate lobe |
| Groove for IVC (right sagittal limb, posterior) | Inferior vena cava | Separates right lobe from caudate lobe |
| Porta hepatis (horizontal crossbar) | Portal vein, hepatic artery proper (2 branches), hepatic ducts, nerves, lymphatics | Connects the two limbs of the H |
Visceral impressions on the liver surface:
- Left side: esophageal impression, gastric impression, omental tuberosity
- Right side: duodenal impression, colic impression (right colic flexure), renal impression (right kidney), suprarenal impression
3. LOBES OF THE LIVER
Morphological (Anatomical) Lobes - based on external surface landmarks
| Lobe | Location | Boundaries |
|---|---|---|
| Right lobe | Largest; right side | To the right of falciform ligament (anterior) / fissure for ligamentum teres (visceral) |
| Left lobe | Smaller; left side | To the left of falciform ligament |
| Quadrate lobe | Anterior visceral surface | Left: fissure for ligamentum teres; Right: fossa for gallbladder; Posterior: porta hepatis. Functionally = LEFT lobe |
| Caudate lobe | Posterior visceral surface | Left: fissure for ligamentum venosum; Right: groove for IVC; Anterior: porta hepatis. Functionally INDEPENDENT |
Important: The traditional 4-lobe division based on external surface features does NOT correspond to the functional (surgical) division of the liver. The functional division is based on the vascular and biliary distribution.
4. COUINAUD'S SEGMENTAL ANATOMY (8 Segments)
This is the most surgically important classification. The liver is divided based on the hepatic arterial, portal venous, and biliary drainage into 8 independent functional segments, each with its own vascular pedicle and bile duct.
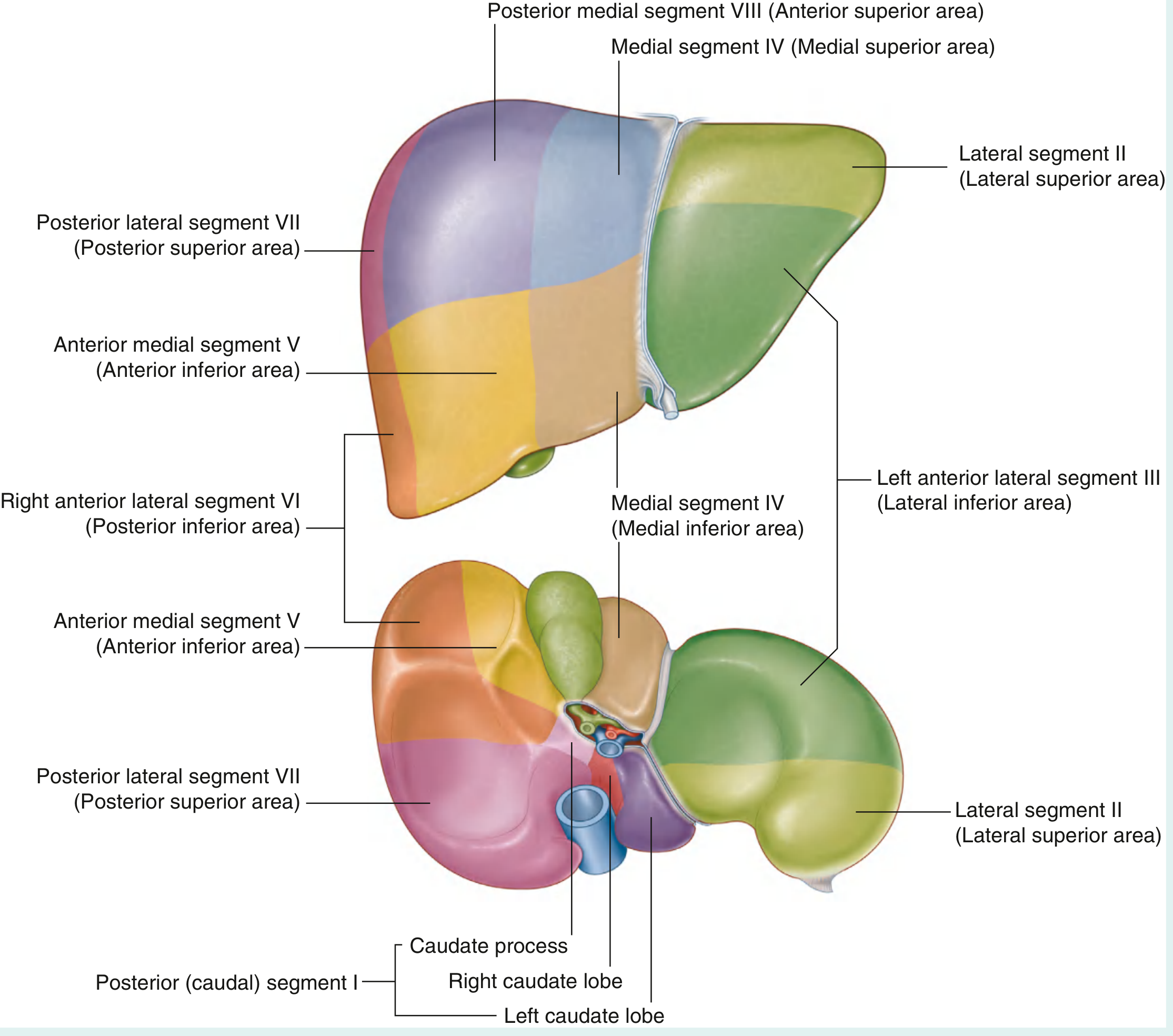
The Principal Plane (Cantlie's Line)
- An imaginary parasagittal line passing through the gallbladder fossa to the inferior vena cava
- Divides the liver into right half and left half of approximately equal size
- The middle hepatic vein lies in this plane
- This is the surgically relevant division - NOT the anatomical right/left lobe division
Segment Numbering (clockwise from caudate lobe):
- Segment I = Caudate lobe (unique - has direct venous drainage into IVC, independent of hepatic veins)
- Segment II = Left lateral superior area (lateral segment, superior)
- Segment III = Left lateral inferior area (lateral segment, inferior)
- Segment IV = Left medial area (quadrate lobe = IVa superior + IVb inferior)
- Segment V = Right anterior inferior area
- Segment VI = Right posterior inferior area
- Segment VII = Right posterior superior area
- Segment VIII = Right anterior superior area
Surgical Resections Based on Segments:
- Right hepatectomy: Segments V + VI + VII + VIII removed; I + II + III + IV remain
- Left hepatectomy: Segments II + III + IV removed; I + V + VI + VII + VIII remain
- Extended right hepatectomy: V + VI + VII + VIII + IV
- Left lateral sectionectomy: II + III
- Individual segment resections possible due to independent pedicles
5. LIGAMENTS OF THE LIVER
| Ligament | From - To | Notes |
|---|---|---|
| Falciform ligament | Liver to anterior abdominal wall and diaphragm | Contains the round ligament (ligamentum teres) in its free inferior margin; derived from ventral mesentery; divides right from left subphrenic recess |
| Coronary ligament | Liver to diaphragm (posterior) | Anterior and posterior layers bound the bare area; where they meet laterally they form triangular ligaments |
| Right triangular ligament | Right lobe to diaphragm | Where anterior and posterior coronary ligaments meet on the right |
| Left triangular ligament | Left lobe to diaphragm | Terminates as the fibrous appendix of the liver |
| Hepatogastric ligament | Liver to lesser curvature of stomach | Part of lesser omentum; contains left and right gastric vessels |
| Hepatoduodenal ligament | Liver to superior duodenum | Part of lesser omentum; the FREE EDGE = right border of lesser omentum = anterior boundary of epiploic foramen (of Winslow); contains the portal triad (portal vein posteriorly, hepatic artery proper on the left, common bile duct on the right) |
| Ligamentum teres (round ligament) | Umbilicus to liver (left branch portal vein) | Remnant of umbilical vein; runs in free margin of falciform ligament; in portal hypertension can recanalize |
| Ligamentum venosum | Left branch portal vein to left hepatic vein / IVC | Remnant of ductus venosus |
The Bare Area
- Located on the posterior diaphragmatic surface of the right lobe
- No peritoneum between liver and diaphragm here
- Bounded anteriorly by anterior coronary ligament, posteriorly by posterior coronary ligament
- Clinically relevant: pathway for spread of subphrenic abscess or retroperitoneal gas
6. VISCERAL SURFACE - PORTA HEPATIS
The porta hepatis (gateway to the liver) is the transverse fissure on the visceral surface forming the crossbar of the H:
Structures ENTERING the liver at the porta hepatis:
- Portal vein (posterior)
- Hepatic artery proper - divides into right and left hepatic arteries
- Nerves (hepatic plexus, from celiac plexus)
Structures LEAVING the liver at the porta hepatis:
- Right hepatic duct + Left hepatic duct (join to form the common hepatic duct)
- Lymphatic vessels
Mnemonic for porta hepatis contents (anterior to posterior within hepatoduodenal ligament):
- Bile duct (right/anterolateral)
- Hepatic artery (left/anteromedial)
- Portal vein (posterior)
- "BD, HA, PV" or "The Patient Has Blood" (PV posterior, HA left, BD right)
7. BLOOD SUPPLY - DUAL CIRCULATION
The liver receives dual blood supply - unique among abdominal organs:
| Source | Vessel | Volume | Oxygen contribution | Notes |
|---|---|---|---|---|
| Venous (nutrient-rich) | Portal vein | ~75% of hepatic blood flow | ~50-60% of O2 | Nutrient-rich from GI tract; formed by SMV + splenic vein behind neck of pancreas |
| Arterial (oxygen-rich) | Hepatic artery proper | ~25% of hepatic blood flow | ~40-50% of O2 | Branch of common hepatic artery from celiac trunk |
Arterial Supply - Origin and Course:
Celiac trunk (from aorta at T12/L1 level) → Common hepatic artery → Hepatic artery proper (after giving off gastroduodenal artery) → divides at porta hepatis into:
- Right hepatic artery → supplies right lobe (segments V-VIII)
- Also gives off cystic artery → gallbladder
- Left hepatic artery → supplies left lobe (segments II-IV)
Common anatomical variation (40% of cases): The right hepatic artery arises from the superior mesenteric artery (replaced right hepatic artery) - surgically critical
Portal Vein Formation:
- Superior mesenteric vein + Splenic vein join posterior to the neck of the pancreas
- The inferior mesenteric vein joins the splenic vein (or SMV junction)
- Portal vein ascends in the hepatoduodenal ligament (posterior to bile duct and hepatic artery)
Venous Drainage:
- Right, Middle, and Left hepatic veins → drain directly into the inferior vena cava (IVC) just below the diaphragm
- The middle hepatic vein often joins the left hepatic vein before entering the IVC
- Caudate lobe drains directly into the IVC via small separate veins (hence preserved in Budd-Chiari syndrome)
- Hepatic veins have NO valves
Clinical Point - Portal Hypertension and Portosystemic Anastomoses:
When portal pressure rises above 12 mmHg, blood is diverted through collateral channels. The 4 main sites of portosystemic anastomoses are:
| Site | Portal side | Systemic side | Clinical result |
|---|---|---|---|
| Lower esophagus | Left gastric (coronary) vein | Azygos / hemiazygos vein | Esophageal varices (most dangerous - bleeding) |
| Umbilicus | Para-umbilical veins (from left portal vein) | Superficial epigastric veins | Caput medusae |
| Rectum | Superior rectal vein (portal) | Middle + inferior rectal veins (systemic) | Hemorrhoids |
| Retroperitoneum | Veins of ascending/descending colon, liver bed | Posterior abdominal wall veins (Retzius veins) | Usually asymptomatic |
8. LYMPHATIC DRAINAGE
- Most abundant lymph production in the body - the liver produces ~25-50% of thoracic duct lymph
- Superficial lymphatics from the diaphragmatic surface → follow hepatic veins → hepatic lymph nodes at porta hepatis → celiac lymph nodes → cisterna chyli → thoracic duct
- Deep lymphatics travel with portal tracts → hepatic lymph nodes
- Some lymphatics from the bare area pass directly through the diaphragm → posterior mediastinal lymph nodes
9. NERVE SUPPLY
- Sympathetic (T7-T10): Via celiac plexus → hepatic plexus → travels with hepatic artery branches; vasomotor function
- Parasympathetic: Vagus nerve (both left and right vagal trunks) → hepatic branches → porta hepatis
- Phrenic nerve (C3-C5): Supplies the capsule of the liver (Glisson's capsule) and diaphragm → referred pain to right shoulder tip (C4 dermatome) in liver/biliary disease
10. MICROSCOPIC ANATOMY (HISTOLOGY)
Three models describe the functional unit of the liver:
Model 1 - Classical (Portal) Lobule
- Hexagonal unit
- Central vein at the center (drains into hepatic vein)
- Portal triads (Glisson triads) at the 6 corners, each containing:
- Interlobular portal vein branch
- Interlobular hepatic artery branch
- Bile ductule
- All enclosed in connective tissue of Glisson's capsule
- Hepatocytes radiate outward in plates (cords) from the central vein
- Sinusoids run between hepatocyte plates from portal triads toward the central vein
- Blood flows: portal triad → sinusoids → central vein
- Bile flows in opposite direction: hepatocytes → bile canaliculi → portal tract bile ductule
Model 2 - Portal Lobule
- Triangle with portal triad at center
- Emphasizes bile flow direction
- Central veins at the 3 corners
Model 3 - Hepatic Acinus (Rappaport's Acinus) - Most Clinically Relevant
- Diamond-shaped unit
- Axis = terminal portal venule (and hepatic arteriole) from the portal tract
- Three concentric zones based on distance from the blood supply:
| Zone | Location | O2 and nutrient supply | Metabolic specialization | Vulnerability |
|---|---|---|---|---|
| Zone 1 (Periportal) | Adjacent to portal triad | Highest O2, most nutrients | Oxidative metabolism, gluconeogenesis, beta-oxidation, bile acid synthesis, urea synthesis | Damaged first in toxic hepatitis (e.g., phosphorus toxicity, eclampsia) |
| Zone 2 (Midzone) | Intermediate | Intermediate | Mixed | Yellow fever |
| Zone 3 (Centrilobular/Perivenular) | Adjacent to central vein | Lowest O2 | Drug metabolism (CYP450), glycolysis, lipogenesis | Damaged first in ischemic/congestive hepatitis, alcohol-induced injury, paracetamol (acetaminophen) toxicity |
Key Cellular Components:
| Cell Type | Location | Function |
|---|---|---|
| Hepatocytes | Form cords/plates; 70-80% of liver mass | Metabolism, bile production, protein synthesis, detoxification |
| Kupffer cells | Lining sinusoids (intravascular) | Liver-resident macrophages; phagocytosis of bacteria, old RBCs, foreign particles; part of the mononuclear phagocyte system (MPS) |
| Stellate cells (Ito cells) | In the Space of Disse | Fat storage (vitamin A); when activated in liver injury, they become myofibroblasts and produce collagen → fibrosis → cirrhosis |
| Sinusoidal endothelial cells | Line sinusoids | Fenestrated (pores ~100 nm), NO basement membrane - allows direct contact between blood and hepatocyte microvilli |
| Pit cells | Attached to sinusoidal endothelium | Natural killer cells of the liver |
| Cholangiocytes | Line bile ducts | Modify bile composition |
Space of Disse
- Perisinusoidal space between sinusoidal endothelium and hepatocyte microvilli
- Contains: stellate cells (Ito cells), plasma (percolates through endothelial fenestrations), reticulin fibers
- Where hepatocyte microvilli project - maximizing exchange surface area
- Hepatocytes have direct contact with blood through this space (no basement membrane barrier)
Bile Canaliculi
- Not separate tubes - formed by grooves between adjacent hepatocytes, sealed by tight junctions
- ~1-2 µm diameter
- Bile flows from zone 3 → zone 1 → bile ductules (canals of Hering) → interlobular bile ducts in portal tracts
- Bile flow is in the opposite direction to blood flow
11. BILIARY SYSTEM ANATOMY
Intrahepatic Biliary System
- Bile canaliculi → canals of Hering (hepatocyte to cholangiocyte transition) → interlobular bile ductules → segmental bile ducts → right and left hepatic ducts
Extrahepatic Biliary System
Right hepatic duct + Left hepatic duct join at the porta hepatis → Common Hepatic Duct (CHD)
CHD + Cystic duct join → Common Bile Duct (CBD)
The CBD (length ~8 cm, diameter normally ≤8 mm):
- Descends in the free edge of hepatoduodenal ligament (to the right of hepatic artery, anterior to portal vein)
- Passes posterior to the superior part of the duodenum
- Passes in a groove on the posterior surface of the head of the pancreas
- Joins the main pancreatic duct (of Wirsung) → forms the hepatopancreatic ampulla (of Vater)
- Opens into the descending (2nd) part of the duodenum at the major duodenal papilla
- Surrounded by the sphincter of Oddi (sphincter of the hepatopancreatic ampulla)
Gallbladder
- Pear-shaped sac, capacity ~50 mL
- Lies in the gallbladder fossa between the right and quadrate lobes
- Parts: Fundus (projects below inferior border, level of tip of 9th costal cartilage at the midclavicular line) → Body (against transverse colon and duodenum) → Neck (with spiral valves of Heister - mucosal spiral folds) → Cystic duct
- Function: receives, concentrates (~10-fold), and stores bile
- Arterial supply: Cystic artery from the right hepatic artery (within the hepatocystic triangle - Calot's triangle)
Calot's Triangle (Hepatocystic Triangle)
Boundaries:
- Superior: inferior surface of liver
- Left: common hepatic duct
- Right: cystic duct
Contents: Cystic artery (right hepatic artery passes through this triangle)
- Surgically important during cholecystectomy
12. RELATIONS OF THE LIVER
Anterior Relations:
- Right and left costal margins
- Xiphoid process and anterior abdominal wall (small strip)
- Diaphragm (above)
Posterior Relations (visceral surface):
- Diaphragm (bare area - no peritoneum)
- Right kidney and right suprarenal gland (renal impression)
- Inferior vena cava (in the IVC groove)
- Esophagus (esophageal impression, left lobe)
- Stomach (gastric impression, left lobe)
- Superior duodenum (duodenal impression)
- Right colic flexure (colic impression)
13. DEVELOPMENT (EMBRYOLOGY)
- The liver develops from the hepatic diverticulum (hepatic bud) - an outgrowth from the ventral wall of the foregut (future duodenum) during week 4
- The diverticulum grows into the septum transversum (mesenchyme between the pericardial cavity and yolk sac stalk)
- Pars hepatica → liver parenchyma and intrahepatic bile ducts
- Pars cystica → gallbladder and cystic duct
- The falciform ligament is derived from the ventral mesentery
- The lesser omentum is derived from the ventral mesentery
- Hematopoiesis occurs in the fetal liver from week 6 to week 28 (then transfers to bone marrow)
- Ductus venosus - fetal vascular channel from the left branch of portal vein to the IVC, bypassing the hepatic sinusoids; closes at birth → ligamentum venosum
- Umbilical vein (left) carries oxygenated blood from placenta to fetus → closes at birth → ligamentum teres (round ligament)
14. CLINICAL ANATOMY - KEY POINTS
| Concept | Detail |
|---|---|
| Hepatorenal recess (Morrison's pouch) | Most dependent peritoneal space in the supine patient; first site of intraperitoneal fluid accumulation; seen on FAST ultrasound in trauma |
| Cantlie's line | Functional division of liver (gallbladder fossa to IVC); site of right hepatectomy |
| Couinaud segments | Foundation for liver resection surgery; individual segments resectable |
| Calot's triangle | Surgical landmark during cholecystectomy; contains cystic artery |
| Referred pain | Right shoulder tip pain in hepatic/biliary disease (via phrenic nerve C4) |
| Caput medusae | Recanalization of paraumbilical veins in portal hypertension |
| Replaced right hepatic artery | 20% of patients - from SMA; must be identified before liver/pancreatic surgery |
| Caudate lobe caudate in Budd-Chiari | Caudate lobe enlarges in Budd-Chiari syndrome because it has independent venous drainage directly to IVC |
| Zone 3 necrosis | Centrilobular; caused by paracetamol toxicity, cardiac failure (congestive hepatopathy), halothane |
| Zone 1 necrosis | Periportal; caused by phosphorus, eclampsia |
| Stellate cell activation | Key step in hepatic fibrosis and cirrhosis; Ito cells → myofibroblasts → collagen deposition |
| Hepatic jaundice | Failure of hepatocytes to conjugate/excrete bilirubin (as distinct from pre-hepatic and post-hepatic) |
Sources: Gray's Anatomy for Students (9780323934237), Color Atlas of Human Anatomy Vol. 2 Internal Organs (9783132424487), Netter's Atlas of Human Anatomy (9780323547048)
That covers the complete anatomy of the liver at textbook depth - from gross anatomy, surfaces, lobes, Couinaud segmentation, ligaments, blood supply, lymphatics, innervation, microanatomy (lobule/acinus/cell types), biliary system, development, and clinical correlates.
Next topics you can request:
- Physiology (bile secretion, bilirubin metabolism, portal circulation, hepatic metabolic functions)
- Biochemistry (urea cycle, bilirubin pathway, fatty acid metabolism, acute phase proteins)
- Pathology (hepatitis, cirrhosis, NAFLD, HCC - Robbins)
- Microbiology (hepatitis viruses A-E)
- Pharmacology (antivirals, drugs for complications of liver disease)
- Medicine (clinical management of liver diseases - Harrison's)
- Surgery (hepatic resection, portal hypertension surgery)
Now physiology
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is the full textbook-level physiology of the liver, synthesized from Guyton & Hall, Ganong's, and Boron & Boulpaep (Medical Physiology).
PHYSIOLOGY OF THE LIVER - Full Textbook-Level Notes
1. FUNCTIONAL ANATOMY (Physiological Perspective)
The human liver contains 50,000 to 100,000 individual lobules. Each lobule is a cylindric structure several mm in length and 0.8-2 mm in diameter. The average transit time for blood across the liver lobule (portal venule to central hepatic vein) is about 8.4 seconds.
Key physiological features:
- Hepatocyte plates are usually one cell thick
- Plasma is in intimate contact with hepatocytes via the Space of Disse
- Sinusoidal endothelium is fenestrated - pores up to 1 µm diameter, NO basement membrane
- This allows even large plasma proteins to freely enter the Space of Disse
- Kupffer cells (resident macrophages) are anchored to the sinusoidal endothelium, projecting into the lumen
- Each hepatocyte is opposed to both a sinusoid and a bile canaliculus simultaneously - different functional domains (basolateral = blood-facing; apical = bile-facing)
2. HEPATIC CIRCULATION AND BLOOD FLOW
Blood Flow Volumes (Guyton & Hall)
| Source | Volume | % of Total | O₂ Contribution |
|---|---|---|---|
| Portal vein | ~1050 mL/min | ~75% | ~50-60% |
| Hepatic artery | ~300 mL/min | ~25% | ~40-50% |
| Total hepatic blood flow | ~1350 mL/min | 27% of resting cardiac output | 100% |
Pressures (Ganong)
- Portal venous pressure: ~10 mmHg (normal range 5-10 mmHg)
- Hepatic venous pressure: ~5 mmHg
- Sinusoidal pressure: lower than portal venous pressure
- Hepatic arteriole pressure: ~90 mmHg at branch level - marked drop across hepatic arterioles maintains sinusoidal low pressure
The normal pressure gradient across the liver (portal vein to hepatic vein) = only ~9 mmHg - indicating normally very low sinusoidal resistance.
Hepatic Arterial Buffer Response (Ganong)
There is an inverse relationship between hepatic arterial flow and portal venous flow:
- When portal flow decreases → adenosine accumulates in periarteriolar space → dilates terminal arterioles → increases hepatic arterial flow
- When portal flow increases → adenosine washed away faster → arterioles constrict → less arterial flow
- Adenosine is produced at a constant metabolic rate; its local concentration governs arteriolar tone
- This buffer response maintains total hepatic blood flow relatively constant despite variation in either input
Sinusoid Recruitment
- Between meals: many sinusoids are collapsed (not perfused)
- After a meal: portal flow increases → "reserve" sinusoids are recruited
- This prevents portal pressure from rising linearly with flow - a protective mechanism
Sympathetic Nervous Control
- Intrahepatic portal vein radicles receive noradrenergic vasoconstrictor fibers (T3-T11 via splanchnic nerves)
- During systemic sympathetic activation (e.g., shock): portal radicles constrict → portal pressure rises → blood is forced through the liver rapidly, bypassing much of hepatic processing → mobilizes ~300-500 mL of blood from the liver reservoir into systemic circulation
- Hepatic artery innervated by hepatic sympathetic plexus
- The liver acts as a blood reservoir - can mobilize large volumes during stress
Hepatic Lymph
- Very high lymph flow - the liver produces 25-50% of total thoracic duct lymph
- The large endothelial pores allow plasma proteins to pass freely into the Space of Disse
- From there, protein-rich fluid enters lymphatic capillaries in the portal tracts
- When hepatic venous pressure rises (e.g., right heart failure, Budd-Chiari): lymph formation overwhelms drainage → ascites (protein-rich in early liver disease)
3. HEPATIC VASCULAR RESERVE FUNCTION
The liver serves as a blood reservoir:
- Normal: contains ~450-500 mL of blood (~10% of total blood volume)
- During heart failure with elevated venous pressure: hepatic blood volume increases greatly - can enlarge the liver significantly (hepatomegaly of cardiac failure)
- In hemorrhage/sympathetic activation: hepatic vasculature constricts, releasing blood into systemic circulation
4. KUPFFER CELLS - RETICULOENDOTHELIAL FUNCTION
The Kupffer cell system of the liver (also called the hepatic macrophage system) is one of the body's most effective means of destroying bacteria:
- Portal blood arriving from the intestine carries large numbers of colon bacteria (especially after meals)
- Kupffer cells destroy virtually 99% of bacteria before portal blood passes through the liver
- This is critical because the portal blood percolates slowly through the sinusoids, exposing all bacteria to the Kupffer cells
- Also phagocytize: old/damaged red blood cells, foreign particles, immune complexes
5. LIVER REGENERATION
From Guyton & Hall - unique to the liver:
The liver has a remarkable capacity to regenerate after injury or partial resection:
- After a partial hepatectomy (removal of up to 70% of liver mass), the remaining liver cells proliferate until the liver approaches its original size
- Growth factors involved: HGF (hepatocyte growth factor), EGF (epidermal growth factor), TGF-α
- Regulatory restraint: once the liver reaches its appropriate size, further cell division stops - controlled by TGF-β (inhibitory signal)
- Regeneration is also triggered by toxic injury to hepatocytes
- The liver is the only solid organ that can fully regenerate to its original mass
6. METABOLIC FUNCTIONS OF THE LIVER
The liver is the central metabolic hub of the body. It processes virtually every class of nutrient arriving via the portal blood. Below are its major metabolic roles.
A. CARBOHYDRATE METABOLISM
| Function | Details |
|---|---|
| Glycogen storage | Stores ~100 g glucose as glycogen; the largest glycogen store in the body |
| Glucose buffer function | Removes excess glucose from portal blood post-meal → stores as glycogen; releases glucose when blood glucose falls - maintains blood glucose in normal range |
| Gluconeogenesis | From amino acids (especially alanine and glutamine), glycerol (from lipolysis), and lactate (Cori cycle); activated when blood glucose falls - zone 1 hepatocytes specialize in this |
| Glycogenolysis | Rapid breakdown of glycogen to glucose; stimulated by glucagon and epinephrine |
| Galactose and fructose conversion | Converts both to glucose-6-phosphate for entry into glycolysis |
| Interconversion | Converts intermediate metabolites of carbohydrate metabolism to fats, amino acids, and other compounds |
Clinical point: In severe liver failure, hypoglycemia is common because both glycogen stores and gluconeogenesis are impaired.
B. FAT (LIPID) METABOLISM
| Function | Details |
|---|---|
| Beta-oxidation of fatty acids | Fatty acids enter hepatocyte mitochondria via carnitine carrier; cleaved into 2-carbon acetyl-CoA units for energy; liver performs beta-oxidation at a very high rate |
| Ketogenesis | When acetyl-CoA production exceeds TCA cycle capacity (starvation, DM): 2 × acetyl-CoA → acetoacetic acid (a ketone body); exported in blood to peripheral tissues for energy; brain and heart can use ketones effectively |
| Synthesis of cholesterol | ~800 mg/day synthesized de novo (HMG-CoA reductase pathway); most cholesterol in the body is liver-synthesized |
| Phospholipid synthesis | Lecithin (phosphatidylcholine) and other phospholipids |
| Lipoprotein synthesis | VLDL, HDL synthesized by liver; LDL derived from VLDL catabolism |
| Triglyceride synthesis | From excess glucose and amino acids (de novo lipogenesis) |
| Fatty acid desaturation | Liver is the primary site for desaturating saturated fatty acids; provides unsaturated fatty acids to all tissues for membrane synthesis |
Hepatic steatosis: In starvation, DM, obesity, alcohol excess - fat accumulates in hepatocytes as triglyceride droplets (fatty liver). This is the earliest histological change in many liver diseases.
C. PROTEIN METABOLISM
This is one of the most critical hepatic functions:
| Function | Details |
|---|---|
| Deamination of amino acids | Removal of amino groups from excess amino acids; NH₃ released → combined with CO₂ → urea (via the urea cycle); urea excreted by kidney |
| Urea synthesis (urea cycle) | The liver is the only organ that performs the complete urea cycle; without this function, NH₃ accumulates → hepatic encephalopathy |
| Transamination | Transfer of amino groups between amino acids to synthesize non-essential amino acids |
| Plasma protein synthesis | Virtually all plasma proteins EXCEPT immunoglobulins are synthesized by the liver |
Plasma Proteins Synthesized by the Liver
| Protein | Normal value | Clinical significance |
|---|---|---|
| Albumin | 3.5-5.0 g/dL | Maintains oncotic pressure; carrier protein; BEST marker of chronic liver synthetic function |
| Prothrombin (Factor II) | - | Vitamin K-dependent; PT/INR rises early in acute liver failure |
| Fibrinogen (Factor I) | 200-400 mg/dL | Coagulation; falls in severe liver disease |
| Factor V | - | NOT Vitamin K-dependent; falls in both hepatocellular failure and DIC |
| Factors VII, IX, X | - | Vitamin K-dependent; fall in cholestasis (fat-soluble vitamin K malabsorption) and liver failure |
| Antithrombin | - | Anticoagulant factor; synthesized by liver |
| Alpha-1 antitrypsin | - | Protease inhibitor; deficiency causes liver disease + emphysema |
| Transferrin | - | Iron transport |
| Ceruloplasmin | - | Copper transport; low in Wilson's disease |
| C-reactive protein (CRP) | - | Acute phase protein; rises in inflammation |
| Alpha-2 macroglobulin | - | Protease inhibitor |
| Haptoglobin | - | Binds free hemoglobin |
| Complement proteins | C3, C4, etc. | Immune defense |
The liver can synthesize up to 50 g of protein per day. Albumin constitutes the largest fraction.
D. DETOXIFICATION AND EXCRETION
The liver detoxifies substances from two sources: gut-derived (portal blood) and systemic circulation.
Phase I Reactions (Guyton / Ganong)
- Oxidation, reduction, hydroxylation
- Mediated by cytochrome P450 enzymes (especially CYP3A4, CYP2D6, CYP2C9)
- Convert lipophilic drugs/toxins to more polar, less active metabolites
- Located predominantly in zone 3 (centrilobular) hepatocytes
Phase II Reactions
- Conjugation (glucuronidation, sulfation, acetylation, glutathione conjugation, methylation)
- Make metabolites water-soluble for biliary or renal excretion
- Example: bilirubin glucuronidation by UGT1A1
Hormonal Inactivation (Guyton)
The liver inactivates/modifies:
- Thyroxine (T4 → T3 conversion and T4 degradation)
- Steroid hormones: estrogens, cortisol, aldosterone, testosterone - converted to water-soluble glucuronide/sulfate conjugates
- Insulin and glucagon: degraded by liver (~50% of portal insulin extracted on first pass)
- Aldosterone: metabolism by liver; in cirrhosis, reduced aldosterone metabolism → hyperaldosteronism → sodium/water retention → ascites and edema
First-Pass Effect
- Drugs absorbed from the gut pass through the portal circulation and are extensively metabolized by the liver before reaching systemic circulation
- This is the hepatic first-pass effect - reduces bioavailability of many orally administered drugs
- Example: propranolol, morphine, lidocaine have extensive first-pass metabolism
- In liver disease: first-pass effect is reduced → higher systemic drug levels → adjust doses
E. VITAMIN AND MINERAL STORAGE
| Substance | Duration of storage |
|---|---|
| Vitamin A | Up to 10 months |
| Vitamin D | 3-4 months |
| Vitamin B12 | At least 1 year (possibly years) |
| Vitamin K | Small amounts (critical for clotting factor synthesis) |
| Iron (as ferritin) | Acts as blood iron buffer; apoferritin + Fe²⁺ → ferritin |
| Copper | Excreted into bile; excess stored (Wilson's disease) |
| Glycogen | Short-term glucose storage |
7. BILE SECRETION
Overview (Guyton & Hall)
- The liver secretes 600-1000 mL/day of bile
- Bile serves two major functions:
- Fat digestion and absorption (emulsification and micelle formation)
- Excretion of waste products (bilirubin, cholesterol, drugs, hormones)
Two Stages of Bile Secretion (Guyton)
Stage 1 - Hepatocyte secretion:
- Hepatocytes secrete bile into bile canaliculi
- Contains: bile acids, cholesterol, lecithin (phosphatidylcholine), bilirubin
- The active secretion of bile acids is the primary osmotic driving force - bile acid secretion drives water flow into canaliculi
- Canalicular bile is transiently hypertonic; tight junctions between hepatocytes are relatively permeable → water, glucose, calcium, amino acids, glutathione, and urea passively enter
Stage 2 - Ductal secretion (cholangiocyte modification):
- As bile flows through ductules and ducts, cholangiocytes modify composition
- They secrete a watery Na⁺ and HCO₃⁻-rich fluid (stimulated by secretin → cAMP → CFTR channels)
- Cholangiocytes also scavenge glucose and amino acids back into circulation (via active transport)
- Gamma-glutamyl transpeptidase (GGT) on the apical membrane of cholangiocytes hydrolyzes glutathione
- Stage 2 can increase total bile volume by up to 100%
- Ductal bile: pH 7.8-8.6; final biliary bicarbonate alkalinity helps neutralize gastric acid in duodenum
Composition of Bile
| Component | Hepatic Duct Bile | Gallbladder Bile |
|---|---|---|
| pH | 7.5-8.6 | 6.0-7.0 |
| Na⁺ (mM) | 141-165 | 220 |
| Bile acids (g/L) | 3-45 | 32 |
| Bilirubin (g/L) | 1-2 | 3 |
| Phospholipids (g/L) | 1.4-8.1 | 34 |
| Cholesterol (g/L) | 1-3.2 | 6.3 |
| HCO₃⁻ (mM) | 12-55 | 19 |
| Proteins (g/L) | 2-20 | 4.5 |
The gallbladder concentrates bile 10-20-fold by actively absorbing Na⁺ (with Cl⁻, water following passively). Capacity = 30-60 mL but stores up to 12 hours of secretion (~450 mL worth of concentrated bile).
Regulation of Bile Secretion
| Stimulus | Mechanism | Effect |
|---|---|---|
| Bile salts (enterohepatic circulation) | Bile salts return to liver in portal blood → re-secreted into bile | Primary driver of total bile secretion; the more bile salts in circulation, the more bile secreted |
| Secretin | Released by S cells of duodenum when acid enters; acts on cholangiocytes → cAMP → CFTR → HCO₃⁻ and water secretion | Up to doubles bile secretion post-meal |
| CCK (cholecystokinin) | Released by I cells when fat and protein enter duodenum; causes gallbladder contraction + Sphincter of Oddi relaxation | Delivers bile to duodenum |
| Vagal stimulation | Weak choleretic effect | Minor role |
| Glucagon | Mild choleretic | Minor role |
Gallbladder Contraction
CCK is the primary stimulus for gallbladder contraction:
- CCK released from I cells (duodenum/jejunum) in response to fat and protein
- Acts on CCK-A receptors on gallbladder smooth muscle → contraction
- Simultaneously relaxes the Sphincter of Oddi
- Vagal nerve also mediates gallbladder contraction (cephalic phase)
- Somatostatin inhibits gallbladder contraction
8. BILE ACIDS AND SALTS
Synthesis
- Liver synthesizes ~6 g/day of bile salts
- Precursor: cholesterol
- Cholesterol → cholic acid (trihydroxy) and chenodeoxycholic acid (dihydroxy) in approximately equal amounts - these are primary bile acids
- Rate-limiting enzyme: 7-alpha-hydroxylase (CYP7A1)
- Primary bile acids are conjugated with glycine (mainly) or taurine (lesser extent) → glycocholic acid, taurocholic acid, glycochenodeoxycholic acid, taurochenodeoxycholic acid
- The sodium and potassium salts of these conjugated bile acids are the bile salts
Secondary Bile Acids
- In the colon, bacteria deconjugate and dehydroxylate primary bile acids:
- Cholic acid → deoxycholic acid
- Chenodeoxycholic acid → lithocholic acid
- These secondary bile acids are partially reabsorbed and re-enter the enterohepatic circulation
Functions of Bile Salts (Guyton)
1. Emulsification:
- Bile salts have detergent properties (amphipathic: hydrophilic + hydrophobic portions)
- Decrease surface tension of fat particles
- Agitation in intestinal tract breaks large fat globules into minute particles (~1 µm diameter)
- Increases total surface area for lipase action by ~1000-fold
2. Micelle Formation (most important):
- Bile salts + fatty acids + monoglycerides + cholesterol + fat-soluble vitamins → mixed micelles
- Micelles are ~3-10 nm diameter water-soluble cylinders
- Transport lipid digestion products from the gut lumen to the brush border of enterocytes
- Without bile salts: up to 40% of ingested fat is lost in stool (steatorrhea)
Enterohepatic Circulation of Bile Salts (Guyton + Ganong)
This is one of the most important physiological circuits in the body:
Liver synthesizes bile salts
↓
Secreted into bile → gallbladder → duodenum
↓
Emulsification and micelle formation in small intestine
↓
~5% diffuse passively from early small intestine (jejunum)
~90% actively reabsorbed from TERMINAL ILEUM (Na⁺-dependent bile salt transporter = ASBT/SLC10A2)
~5% lost in feces (replaced by new synthesis)
↓
Absorbed bile salts enter PORTAL BLOOD
↓
Liver extracts virtually 100% on first pass
↓
Re-secreted into bile (recycled)
Key facts:
- Total bile salt pool = ~2.5 g
- Recycled 6-10 times per day
- Total bile salt flux per day = ~15-25 g
- If terminal ileum is diseased (Crohn's disease) or resected: bile salt malabsorption → steatorrhea, fat-soluble vitamin deficiency, cholerheic diarrhea
- Cholestyramine (bile acid sequestrant) interrupts this cycle → forces liver to synthesize new bile acids from cholesterol → lowers serum cholesterol
9. BILIRUBIN METABOLISM AND EXCRETION
This is one of the most tested topics in hepatology physiology.
Step 1 - Formation of Bilirubin (in Macrophages)
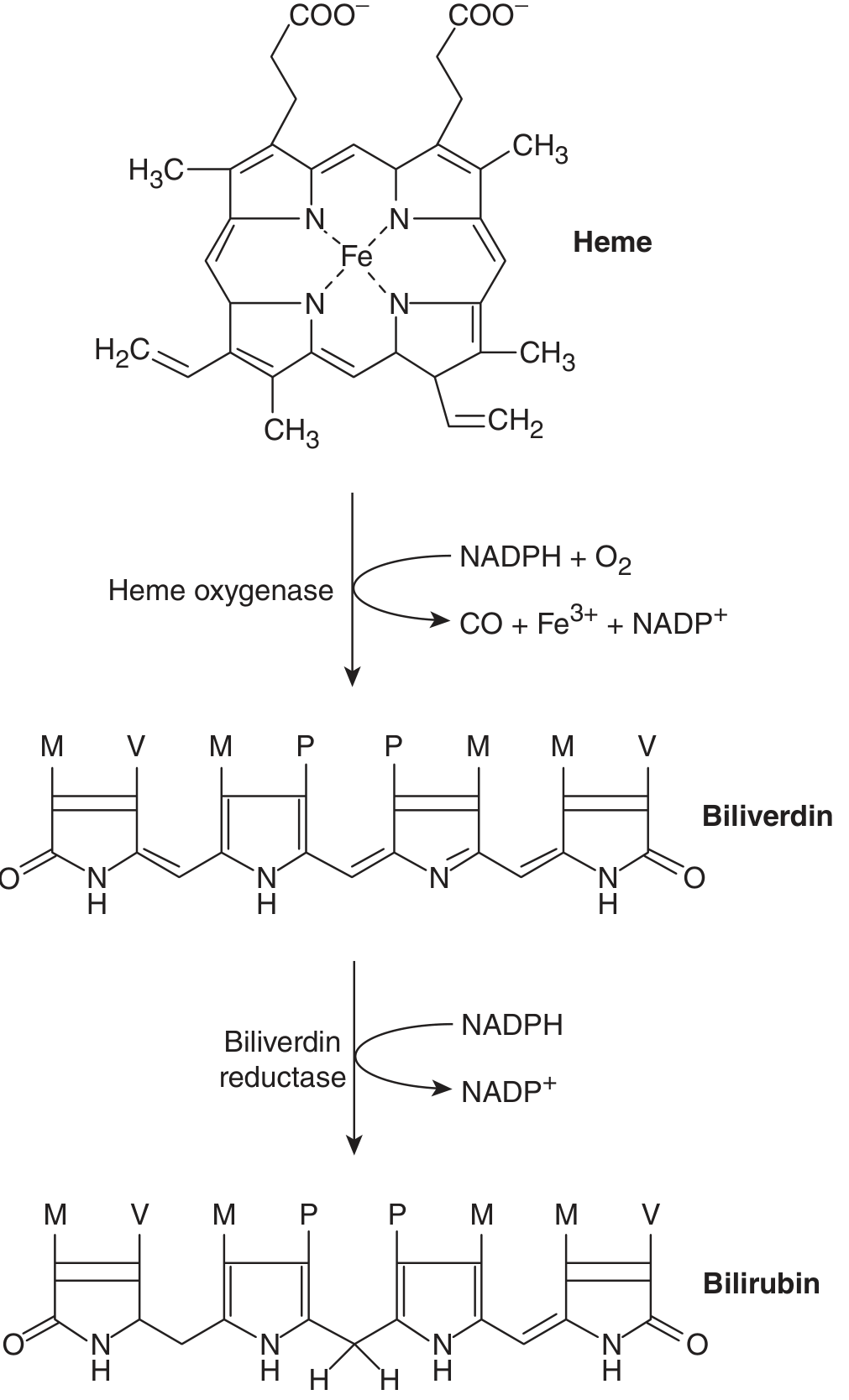
Source of bilirubin:
- 65-80% from breakdown of hemoglobin of senescent red blood cells (RBC lifespan = 120 days)
- Remaining 20-35% from: myoglobin, cytochromes, ineffective erythropoiesis ("early labeled peak")
Process:
- Senescent RBCs → phagocytized by macrophages (spleen, bone marrow, Kupffer cells)
- Hemoglobin → split into globin + heme
- Heme ring opened by heme oxygenase (NADPH + O₂ required) → biliverdin (green pigment) + free iron (Fe³⁺) + CO
- Biliverdin reductase (NADPH required) → bilirubin (yellow pigment)
- Free iron bound to transferrin for recycling
Step 2 - Transport in Blood
- Bilirubin formed in macrophages is lipophilic and insoluble in water
- Binds strongly but reversibly to albumin in plasma → "albumin-bound unconjugated bilirubin"
- Also called: unconjugated bilirubin = indirect bilirubin (reacts only after addition of alcohol in van den Bergh test)
- Normal plasma concentration: 0.1-0.5 mg/dL (mostly unconjugated)
- Does NOT appear in urine (albumin-bound, cannot be filtered by kidney)
- TOXIC to brain when unbound (in neonates: kernicterus when free unconjugated bilirubin crosses blood-brain barrier)
Step 3 - Hepatic Uptake
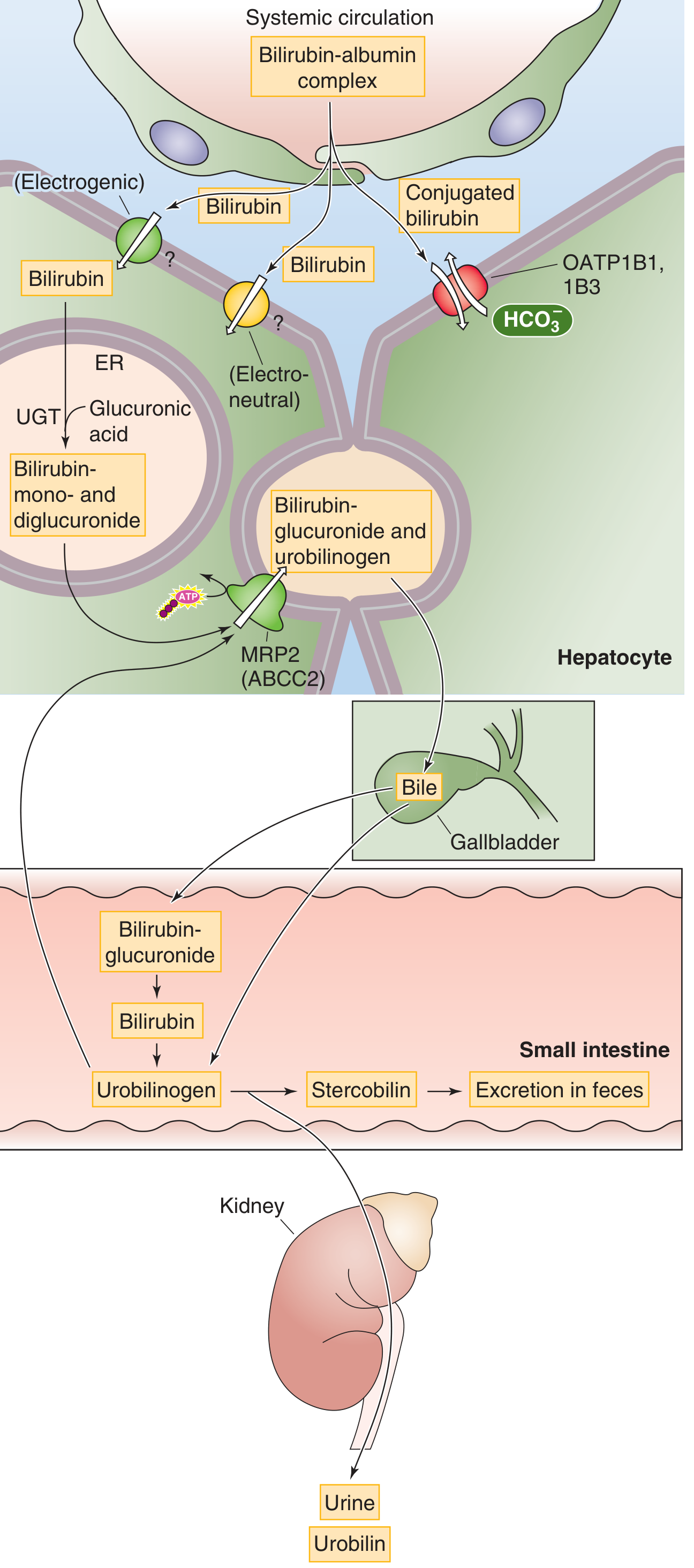
- Albumin-bilirubin complex enters Space of Disse via sinusoidal fenestrations
- Bilirubin dissociates from albumin
- Uptake across basolateral (sinusoidal) membrane by:
- OATP1B1 and OATP1B3 (Organic Anion Transporting Polypeptides) - primary carriers
- Additional passive/facilitated mechanisms
- Inside hepatocyte: bilirubin bound to ligandin (Y protein / glutathione-S-transferase) → prevents back-diffusion
Step 4 - Conjugation (in Hepatocyte ER)
- UDP-glucuronyl transferase (UGT1A1) in smooth ER
- Bilirubin + UDP-glucuronic acid → bilirubin glucuronide
- ~80% → bilirubin diglucuronide (predominant conjugated form)
- ~10% → bilirubin monoglucuronide
- ~10% → bilirubin sulfate and other conjugates
- Conjugated bilirubin = direct bilirubin (reacts directly in van den Bergh test without alcohol)
- Now water-soluble, non-toxic, can be filtered by kidney
Step 5 - Canalicular Secretion
- Conjugated bilirubin secreted from hepatocyte into bile canaliculus by MRP2 (ABCC2) - ATP-dependent (active transport)
- This is the rate-limiting step in hepatic bilirubin handling
- Defect in MRP2 → Dubin-Johnson syndrome (conjugated hyperbilirubinemia)
Step 6 - Intestinal Metabolism (Enterohepatic Bilirubin Cycle)
- Conjugated bilirubin arrives in terminal ileum and colon
- Intestinal bacteria (beta-glucuronidases) → deconjugate bilirubin → free bilirubin
- Further bacterial reduction → urobilinogen (colorless, highly soluble)
- Fate of urobilinogen:
- ~80-90% remains in colon → oxidized to stercobilin → excreted in feces (gives stool its brown color)
- ~10-20% reabsorbed from colon/terminal ileum → enters portal blood → most re-excreted by liver (enterohepatic circulation) → ~5% escapes to systemic circulation → kidney → urine → oxidized to urobilin (yellow color of urine)
Summary Table: Types of Jaundice
| Type | Cause | Unconjugated | Conjugated | Urine bilirubin | Urine urobilinogen | Stool color |
|---|---|---|---|---|---|---|
| Pre-hepatic (Hemolytic) | Excess RBC destruction; liver overwhelmed | ↑↑ | Normal | Absent (albumin-bound) | ↑↑ | Normal/dark |
| Hepatic (Hepatocellular) | Hepatitis, cirrhosis; failure of uptake/conjugation/excretion | ↑ | ↑ | Present (conjugated leaks) | ↓ or normal | Pale |
| Post-hepatic (Obstructive/Cholestatic) | CBD stone, carcinoma head of pancreas; bile flow blocked | Normal | ↑↑ | Present (dark urine) | Absent (no bile reaching gut) | Pale/clay-colored |
Jaundice becomes clinically visible when total serum bilirubin > 1.5-3 mg/dL (Guyton: skin yellows at ~3× normal = 1.5 mg/dL; Medical Physiology: 1.5-3 mg/dL).
Important Inherited Disorders of Bilirubin Metabolism
| Disorder | Defect | Type of Hyperbilirubinemia | Notes |
|---|---|---|---|
| Gilbert's syndrome | Reduced UGT1A1 activity (~30% of normal) | Unconjugated | Benign; worsens with fasting/illness; no treatment needed |
| Crigler-Najjar type I | Complete absence of UGT1A1 | Unconjugated (severe) | Kernicterus; requires liver transplant or phototherapy |
| Crigler-Najjar type II | Partial UGT1A1 deficiency | Unconjugated (moderate) | Responds to phenobarbital |
| Dubin-Johnson syndrome | Defective MRP2 (ABCC2) - canalicular secretion | Conjugated | Benign; black liver (melanin pigment); urinary coproporphyrin I ↑ |
| Rotor syndrome | Defective OATP1B1/1B3 (hepatocyte uptake) | Conjugated | Benign; no liver pigment |
10. CHOLESTEROL AND GALLSTONE FORMATION
Hepatic Cholesterol Secretion (Ganong/Guyton)
- The liver secretes cholesterol into bile in a micellar solution together with bile acids and phosphatidylcholine (lecithin)
- The ratio in canalicular bile: bile acids : phosphatidylcholine : cholesterol ≈ 10 : 3 : 1
- If this ratio is disturbed (↑ cholesterol or ↓ bile acids), cholesterol precipitates → cholesterol gallstones (80% of gallstones in Western populations)
Gallstone Risk
- Deviations from the bile acid:lecithin:cholesterol ratio (above line ABC on Admirand-Small triangle) → cholesterol saturation → nucleation → gallstone formation
- Risk factors: obesity, rapid weight loss, prolonged fasting, ileal disease (reduced bile salt pool), female sex, OCPs, fibrates
11. PORTAL HYPERTENSION AND HEPATIC ENCEPHALOPATHY
Portal Hypertension (Boron & Boulpaep / Ganong)
- Normal portal pressure: ~10 mmHg
- Portal hypertension defined as: portal pressure > 12 mmHg (or hepatic venous pressure gradient > 10 mmHg)
- When sinusoidal resistance rises (cirrhosis, fibrosis): portal pressure rises
- Because hepatic sinusoids are highly permeable (low reflection coefficient), even small increases in pressure lead to massive lymph formation and ascites
- In cirrhosis: hardening and fibrosis of liver → resistance to sinusoidal flow → backup of portal pressure → all portosystemic collaterals open up (see anatomy section)
Hepatic Encephalopathy (Ganong)
- In portal hypertension: blood bypasses the liver via collaterals → gut-derived toxins (especially ammonia/NH₃) enter systemic circulation
- Ammonia is the primary toxin: derived from gut bacteria deaminating amino acids; normally detoxified by liver → urea (urea cycle)
- In liver failure: urea cycle fails → NH₃ accumulates → brain toxicity
- Other toxins: mercaptans, short-chain fatty acids, false neurotransmitters (octopamine), GABA-like substances
- Treatment: lactulose (traps NH₃ as NH₄⁺ in gut lumen) + rifaximin (non-absorbed antibiotic reduces gut bacteria) + restrict dietary protein
12. HEPATIC REGULATION OF BLOOD GLUCOSE - THE "GLUCOSE BUFFER" FUNCTION
| State | Hepatic Response | Mechanism |
|---|---|---|
| Post-meal (fed state) | Removes ~80% of portal glucose; stores as glycogen | Insulin stimulates glycogen synthase; portal glucose concentration directly stimulates hepatic glucose uptake (glucose sensor) |
| Fasting (2-6 hrs) | Releases glucose from glycogen (glycogenolysis) | Glucagon activates glycogen phosphorylase via cAMP |
| Prolonged fasting (>12 hrs) | Glucose production by gluconeogenesis | Glucagon, cortisol, epinephrine; substrates: alanine (from muscle), lactate (Cori cycle), glycerol (from fat breakdown) |
| Liver failure | Inability to buffer → severe hypoglycemia after meals or during fasting | Loss of glucose buffer function |
13. LIVER FUNCTION TESTS - PHYSIOLOGICAL BASIS
| Test | What it measures | Reflects |
|---|---|---|
| ALT (Alanine aminotransferase) | Hepatocyte enzyme | Hepatocyte damage/necrosis (most specific for liver) |
| AST (Aspartate aminotransferase) | Hepatocyte + mitochondrial enzyme | Hepatocyte damage (less specific - also in heart, muscle) |
| ALP (Alkaline phosphatase) | Canalicular membrane enzyme | Cholestasis (bile duct disease, biliary obstruction) |
| GGT (Gamma-glutamyl transpeptidase) | Cholangiocyte enzyme; induced by alcohol | Cholestasis, alcohol use; confirms ALP is hepatic origin |
| Total bilirubin | All bilirubin in plasma | Overall bilirubin handling |
| Direct bilirubin | Conjugated bilirubin | Conjugation completed; elevated in cholestasis and hepatocellular disease |
| Albumin | Serum albumin level | Chronic hepatic synthetic function (half-life = 20 days) |
| PT/INR | Prothrombin time | Acute hepatic synthetic function (clotting factors: half-life hours-days) |
| Ammonia | Serum NH₃ | Urea cycle function; correlates with encephalopathy |
14. HEPATIC FIRST-PASS METABOLISM AND PHARMACOKINETICS
- The liver is the primary site of drug metabolism in the body
- Drug oral absorption → portal vein → liver → extensive first-pass hepatic metabolism → systemic circulation
- Bioavailability = fraction of oral dose reaching systemic circulation after first-pass metabolism
- High first-pass drugs: propranolol (bioavailability ~25%), morphine (~25%), aspirin, lidocaine, nitrates, verapamil, statins (some)
- Hepatic blood flow-limited drugs (flow-dependent): Lidocaine, propranolol - extraction so complete that clearance depends on hepatic blood flow, not enzyme activity
- Enzyme-limited (capacity-dependent) drugs: Warfarin, phenytoin - extraction limited by enzyme capacity/affinity
- In liver disease: reduced first-pass → increased bioavailability of many drugs → dose reduction needed
- In portal hypertension: portosystemic shunting → drugs bypass liver entirely → even greater bioavailability
Sources: Guyton and Hall Textbook of Medical Physiology (9780443111013), Ganong's Review of Medical Physiology 26th Ed (9781260122404), Medical Physiology - Boron & Boulpaep (9780323319737)
That covers the complete physiology of the liver - from hepatic microcirculation, metabolic functions (carbohydrate, fat, protein), bile secretion, bile salt chemistry and enterohepatic circulation, bilirubin metabolism and jaundice classification, cholesterol/gallstone formation, portal hypertension, hepatic encephalopathy, and pharmacokinetic implications.
Next topic you can request:
- Biochemistry (urea cycle in detail, bilirubin pathway at molecular level, fatty acid metabolism, ketone bodies, Harper's + Lippincott)
- Pathology (hepatitis, cirrhosis, NAFLD, HCC - Robbins)
- Microbiology (Hepatitis A-E viruses in depth)
- Pharmacology (antivirals, lactulose, rifaximin, terlipressin, UDCA, statins and liver)
- Medicine (clinical management - Harrison's)
- Surgery (hepatic resection, portal hypertension surgery)
Biochemistry
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is the complete textbook-level biochemistry of the liver, synthesized from Harper's Illustrated Biochemistry (32nd Ed), Lippincott Illustrated Reviews: Biochemistry (8th Ed), and Basic Medical Biochemistry: A Clinical Approach (6th Ed).
BIOCHEMISTRY OF THE LIVER - Full Textbook-Level Notes
1. THE LIVER AS THE CENTRAL METABOLIC ORGAN
The liver is interposed between the general circulation and the digestive tract. It receives ~20% of cardiac output - via the portal vein (nutrient-rich, gut-absorbed) and the hepatic artery (oxygenated systemic blood). Its relatively large size (~3% of body weight) allows extended residence time for nutrients to be metabolized and toxic agents to be detoxified before reaching other organs.
The hepatocyte is the master metabolic cell. It performs:
- Carbohydrate metabolism (glycolysis, gluconeogenesis, glycogen synthesis/breakdown)
- Lipid metabolism (beta-oxidation, ketogenesis, fatty acid synthesis, cholesterol/lipoprotein synthesis)
- Protein/amino acid metabolism (transamination, deamination, urea cycle, plasma protein synthesis)
- Bilirubin metabolism
- Detoxification (Phase I and Phase II drug metabolism)
- Bile acid synthesis
- Vitamin and mineral storage
Sequential transport steps within the hepatocyte: uptake → intracellular binding/sequestration → metabolism → sinusoidal secretion → biliary excretion.
2. CARBOHYDRATE METABOLISM IN THE LIVER
A. Glycolysis and Glucokinase
- Hepatocytes express glucokinase (hexokinase IV) - a high-Km, high-capacity, non-saturable enzyme for glucose
- Unlike hexokinase in other tissues, glucokinase is NOT inhibited by its product glucose-6-phosphate
- This allows the liver to act as a glucose buffer: after a meal, when portal glucose is high, the liver phosphorylates and traps large amounts of glucose
- Glucokinase is induced by insulin and repressed by fasting
B. Glycogen Synthesis (Glycogenesis)
- Glucose-6-phosphate → glucose-1-phosphate (phosphoglucomutase) → UDP-glucose (UDP-glucose pyrophosphorylase) → glycogen (glycogen synthase)
- Glycogen synthase is the key regulatory enzyme: activated by insulin (dephosphorylation) and glucose-6-phosphate; inactivated by glucagon/epinephrine (phosphorylation via PKA)
- Branching enzyme creates alpha-1,6-glycosidic branch points every 8-10 residues
- Liver glycogen = ~100 g (muscle = ~400 g); liver glycogen primarily for maintaining blood glucose; muscle glycogen for local use only
C. Glycogenolysis
- Glycogen phosphorylase (rate-limiting): cleaves alpha-1,4 bonds → glucose-1-phosphate
- Activated by: glucagon (via cAMP → PKA → phosphorylase kinase → glycogen phosphorylase), epinephrine, calcium (in muscle)
- Debranching enzyme handles the alpha-1,6 branch points → releases free glucose
- Glucose-1-phosphate → glucose-6-phosphate → glucose-6-phosphatase (liver only, NOT muscle) → free glucose exported to blood
- Muscle lacks glucose-6-phosphatase → cannot export glucose (muscle glycogen is for local use)
D. Gluconeogenesis
- Synthesis of glucose from non-carbohydrate precursors - occurs almost exclusively in liver and kidney cortex
- Substrates:
- Lactate (from anaerobic glycolysis in RBCs, muscles) → pyruvate → glucose (Cori cycle)
- Alanine (from muscle protein catabolism) → pyruvate → glucose (glucose-alanine cycle)
- Glycerol (from lipolysis of triglycerides in adipose tissue) → glycerol-3-phosphate → DHAP → glucose
- Glucogenic amino acids (all except leucine and lysine)
- Propionate (from odd-chain fatty acid oxidation)
4 irreversible steps of gluconeogenesis (bypass the irreversible glycolytic reactions):
| Glycolytic reaction (irreversible) | Gluconeogenic bypass enzyme | Subcellular location |
|---|---|---|
| Pyruvate kinase (PEP → pyruvate) | Pyruvate carboxylase (pyruvate → OAA) then PEPCK (OAA → PEP) | Mitochondria then cytosol |
| Phosphofructokinase-1 (F6P → F1,6-BP) | Fructose-1,6-bisphosphatase (F1,6-BP → F6P) | Cytosol |
| Hexokinase/Glucokinase (Glucose → G6P) | Glucose-6-phosphatase (G6P → glucose) | ER membrane (liver/kidney only) |
Regulation of gluconeogenesis:
- Activated by: glucagon (↑cAMP → ↑PEPCK expression, ↑fructose-2,6-bisphosphatase → ↓F2,6-BP → relieves PFK-1 activation → allows F1,6-BPase to act), cortisol (induces PEPCK), fasting/starvation, high ATP:ADP ratio
- Inhibited by: insulin (↓PEPCK transcription), high AMP (inhibits PEPCK), high F2,6-bisphosphate
Biotin (as cofactor for pyruvate carboxylase) is required for gluconeogenesis.
3. LIPID METABOLISM IN THE LIVER
A. Fatty Acid Catabolism - Beta-Oxidation
Transport of Long-Chain Fatty Acids into Mitochondria - The Carnitine Shuttle (Lippincott):
- Fatty acid + CoA + ATP → Fatty acyl-CoA (catalyzed by fatty acyl-CoA synthetase/thiokinase; outer mitochondrial membrane)
- Fatty acyl-CoA + carnitine → acylcarnitine + CoA (catalyzed by CPT-I = carnitine palmitoyltransferase I; outer mitochondrial membrane)
- CPT-I is the rate-limiting step and the key regulatory site of beta-oxidation
- CPT-I is inhibited by malonyl-CoA (the first intermediate of fatty acid synthesis) - this prevents simultaneous synthesis and oxidation of fatty acids
- Acylcarnitine transported across inner mitochondrial membrane by carnitine-acylcarnitine translocase (antiporter, exchanges for free carnitine)
- Acylcarnitine + CoA → acyl-CoA + carnitine (catalyzed by CPT-II; inner membrane)
- Acyl-CoA enters beta-oxidation spiral in mitochondrial matrix
The Beta-Oxidation Spiral (for saturated even-chain fatty acid):
Each cycle removes a 2-carbon unit as acetyl-CoA and produces 1 NADH + 1 FADH₂:
| Step | Enzyme | Reaction |
|---|---|---|
| 1 | Acyl-CoA dehydrogenase (FAD-dependent) | Acyl-CoA → trans-enoyl-CoA + FADH₂ |
| 2 | Enoyl-CoA hydratase | trans-enoyl-CoA + H₂O → L-3-hydroxyacyl-CoA |
| 3 | L-3-hydroxyacyl-CoA dehydrogenase (NAD⁺-dependent) | L-3-hydroxy-acyl-CoA → 3-ketoacyl-CoA + NADH |
| 4 | Thiolase (acyl-CoA acyltransferase) | 3-ketoacyl-CoA + CoA → acetyl-CoA + shortened acyl-CoA |
Energy yield from palmitate (C16:0) = 7 cycles:
- 7 cycles × (1 FADH₂ + 1 NADH) = 7 FADH₂ + 7 NADH
- 8 acetyl-CoA → 8 × 10 ATP = 80 ATP
- Total = 7(1.5) + 7(2.5) + 80 - 2 (for activation) = 106 ATP net
Odd-chain fatty acids → last cycle yields propionyl-CoA → propionyl-CoA carboxylase (biotin) → methylmalonyl-CoA → succinyl-CoA (vitamin B12) → enters TCA cycle. This is a source of glucose (propionate is gluconeogenic).
Unsaturated fatty acids: require enoyl-CoA isomerase (for cis double bonds at odd carbons) and 2,4-dienoyl-CoA reductase (for even-positioned double bonds).
B. Ketone Body Synthesis (Ketogenesis) - Occurs Exclusively in Liver Mitochondria
When acetyl-CoA production from beta-oxidation exceeds TCA cycle capacity (fasting, starvation, DM Type 1, high-fat diet):
2 Acetyl-CoA → Acetoacetyl-CoA [thiolase]
Acetoacetyl-CoA + Acetyl-CoA → HMG-CoA [HMG-CoA synthase] ← rate-limiting step
HMG-CoA → Acetoacetate + Acetyl-CoA [HMG-CoA lyase]
Acetoacetate → β-Hydroxybutyrate [β-hydroxybutyrate dehydrogenase; NADH-dependent]
Acetoacetate → Acetone (spontaneous, non-enzymatic decarboxylation; minor)
Key points:
- The liver produces ketone bodies but cannot use them (lacks succinyl-CoA:acetoacetate CoA transferase = thiophorase)
- Ketone bodies are exported via blood to brain, heart, skeletal muscle, kidney for use as fuel
- In prolonged starvation, brain adapts to use ketone bodies as primary fuel (up to 70%), sparing glucose/protein
- β-Hydroxybutyrate is the predominant ketone body in starvation
- Acetone is volatile → excreted in breath (fruity smell in diabetic ketoacidosis)
- Clinical: Diabetic Ketoacidosis (DKA): Severe insulin deficiency → uncontrolled lipolysis → massive beta-oxidation → massive ketogenesis → ketoacidosis
Regulation of ketogenesis:
- Malonyl-CoA levels control CPT-I: high malonyl-CoA (active lipogenesis, fed state) → ↓CPT-I → ↓beta-oxidation → ↓ketogenesis
- In fasting/DM: insulin ↓ → malonyl-CoA ↓ (ACC inhibited by glucagon) → CPT-I active → ↑beta-oxidation → ↑ketogenesis
C. Fatty Acid Synthesis (De Novo Lipogenesis)
Occurs in cytosol, primarily in liver (and adipose tissue, lactating mammary gland):
Acetyl-CoA must first exit mitochondria as citrate (citrate shuttle):
- Acetyl-CoA + OAA → citrate (in mitochondria) → exported to cytosol → cleaved by ATP-citrate lyase → acetyl-CoA + OAA in cytosol
Steps:
- Acetyl-CoA + CO₂ + ATP → Malonyl-CoA catalyzed by acetyl-CoA carboxylase (ACC) - the committed, rate-limiting step
- Requires biotin cofactor
- Activated by: citrate (allosteric), insulin (dephosphorylation)
- Inhibited by: palmitoyl-CoA (product inhibition), glucagon/epinephrine (phosphorylation via AMP kinase), malonyl-CoA itself at high concentrations
- Fatty acid synthase complex (FAS) - a multifunctional enzyme in eukaryotes with 7 enzymatic activities:
- Catalyzes sequential addition of 2-carbon malonyl units onto acetyl-CoA starter unit
- Each elongation cycle: reduction (NADPH) → dehydration → reduction (NADPH)
- Product: Palmitate (C16:0) - the primary product of FAS in humans
- Overall reaction: Acetyl-CoA + 7 Malonyl-CoA + 14 NADPH + H⁺ → Palmitate + 7CO₂ + 14 NADP⁺ + 8 CoA + 6H₂O
NADPH requirement - sources:
- Pentose phosphate pathway (main source, also liver)
- Malic enzyme (malate → pyruvate + CO₂ + NADPH)
Elongation beyond palmitate: Occurs in endoplasmic reticulum (elongases) and mitochondria.
Desaturation: Hepatic desaturases (Δ9, Δ6, Δ5) introduce double bonds; cannot introduce double bonds beyond Δ9 → linoleic acid (18:2, n-6) and alpha-linolenic acid (18:3, n-3) are essential fatty acids - must come from diet.
D. Cholesterol Synthesis
Occurs in liver (and intestine, adrenal cortex, gonads) - liver is the primary site.
From acetyl-CoA → cholesterol (30-step pathway, 5 stages):
Stage 1 - Synthesis of HMG-CoA (in cytosol)
- 3 Acetyl-CoA → HMG-CoA (via thiolase and HMG-CoA synthase)
Stage 2 - HMG-CoA → Mevalonate (RATE-LIMITING STEP)
- Enzyme: HMG-CoA reductase (ER membrane)
- Reaction: HMG-CoA + 2 NADPH → Mevalonate + CoA
- This is the target of statins (competitive inhibitors of HMG-CoA reductase)
Stage 3 - Mevalonate → Isopentenyl pyrophosphate (IPP, C5)
- Requires 3 ATP; produces CO₂
Stage 4 - IPP condensation → Squalene (C30)
- 2 × IPP → GPP (geranyl-PP, C10) → FPP (farnesyl-PP, C15) → squalene (C30, by squalene synthase, requires NADPH)
Stage 5 - Squalene → Cholesterol
- Squalene → lanosterol (by squalene epoxidase + cyclase) → cholesterol (multiple steps, ~20 reactions)
Regulation of Cholesterol Synthesis (Harper's):
- HMG-CoA reductase - master regulatory enzyme:
- Transcriptional: regulated by SREBP (sterol response element binding protein) system:
- When intracellular cholesterol ↓: SCAP (SREBP cleavage-activating protein) escorts SREBP to Golgi → cleaved by proteases → activated SREBP enters nucleus → activates HMG-CoA reductase gene transcription
- When cholesterol ↑: INSIG proteins retain SCAP-SREBP in ER → no gene transcription
- Post-translational: cholesterol promotes ubiquitination and proteasomal degradation of HMG-CoA reductase
- Phosphorylation: AMP kinase phosphorylates and inactivates HMG-CoA reductase (like ACC); insulin activates phosphatase → activates HMG-CoA reductase
- Induced by: insulin, thyroid hormone
- Inhibited by: statins, glucagon, glucocorticoids, bile acids (via FXR)
- Transcriptional: regulated by SREBP (sterol response element binding protein) system:
Cholesterol derivatives in liver:
- Bile acids (main route of cholesterol excretion)
- Steroid hormones (in adrenal, gonads - not liver)
- Vitamin D3 precursor
E. Bile Acid Synthesis (Harper's + Ganong)
Primary bile acid synthesis - occurs exclusively in liver:
- Cholesterol → 7α-hydroxycholesterol (enzyme: CYP7A1 = 7-alpha-hydroxylase - rate-limiting step)
- Two pathways diverge → cholic acid (trihydroxy, 3α,7α,12α-OH) and chenodeoxycholic acid (dihydroxy, 3α,7α-OH)
- These are conjugated with glycine (mainly) or taurine → glycocholic acid, taurocholic acid, glycochenodeoxycholic acid, taurochenodeoxycholic acid = primary bile salts secreted into bile
Secondary bile acids (formed by intestinal bacteria):
- Cholic acid → deoxycholic acid (by 7-dehydroxylation)
- Chenodeoxycholic acid → lithocholic acid (by 7-dehydroxylation)
Regulation of bile acid synthesis (Harper's):
- Feedback regulation via FXR (Farnesoid X Receptor = nuclear bile acid-binding receptor):
- When bile acid pool is large (abundant bile acids in enterohepatic circulation) → bile acids bind FXR → FXR activated → suppresses CYP7A1 transcription → ↓bile acid synthesis
- When bile acid pool is depleted → FXR not activated → ↑CYP7A1 transcription → ↑bile acid synthesis
- Chenodeoxycholic acid is most potent FXR activator
- CYP7A1 also upregulated by: cholesterol (substrate availability), insulin (fed state)
- CYP7A1 downregulated by: glucagon, glucocorticoids, thyroid hormone (species-dependent)
F. Lipoprotein Synthesis and Metabolism
The liver is the primary site of endogenous lipoprotein production:
| Lipoprotein | Synthesized by | Function | Apolipoprotein |
|---|---|---|---|
| VLDL | Liver | Transport endogenous TG from liver to peripheral tissues | ApoB-100, ApoC-II, ApoE |
| IDL | Derived from VLDL in blood | Intermediate - can be taken up by liver or converted to LDL | ApoB-100, ApoE |
| LDL | Derived from IDL (by hepatic lipase) | Delivers cholesterol to peripheral tissues and liver | ApoB-100 |
| HDL (nascent) | Liver + intestine | Reverse cholesterol transport (periphery → liver) | ApoA-I |
| Chylomicrons | Intestine only | Transport dietary (exogenous) TG and cholesterol | ApoB-48 |
VLDL assembly (in liver):
- TG + Phospholipid + Cholesterol + ApoB-100 → VLDL (assembled in ER, processed in Golgi)
- MTP (microsomal triglyceride transfer protein) is essential for VLDL assembly - transfers lipids onto ApoB-100
- VLDL secreted into sinusoidal blood → processed in circulation by lipoprotein lipase (LPL) on capillary walls (activated by ApoC-II) → TG hydrolyzed → fatty acids released to tissues → VLDL → IDL → LDL
LDL Receptor pathway (Brown and Goldstein - Nobel 1985):
- LDL binds LDL receptor (recognizes ApoB-100) → receptor-mediated endocytosis → lysosomal degradation → cholesterol released inside cell → suppresses SREBP → ↓HMG-CoA reductase + ↓LDL receptor synthesis + ↑ACAT (stores excess cholesterol as cholesteryl ester)
- Statins → ↓HMG-CoA reductase → ↓intracellular cholesterol → ↑SREBP activation → ↑LDL receptor expression → ↓plasma LDL
Reverse Cholesterol Transport:
- HDL (ApoA-I) accepts free cholesterol from peripheral cells via ABCA1 transporter
- LCAT (lecithin-cholesterol acyltransferase) activated by ApoA-I: esterifies cholesterol → cholesteryl ester stored in HDL core
- HDL-cholesterol transported to liver via SR-BI receptor (selective uptake, not endocytosis) or transferred to LDL/VLDL by CETP (cholesteryl ester transfer protein)
- In liver: cholesterol excreted as bile acids or free cholesterol in bile
4. PROTEIN AND AMINO ACID METABOLISM IN THE LIVER
A. Transamination (Aminotransferases)
Transamination is the first step in amino acid catabolism:
- Transfer of an α-amino group from an amino acid to an α-keto acid
- Reaction: Amino acid₁ + α-keto acid₂ ⇌ α-keto acid₁ + Amino acid₂
- Freely reversible (equilibrium constant ≈ 1)
- All aminotransferases require pyridoxal phosphate (PLP, Vitamin B₆) as cofactor (covalently bound Schiff base)
- Pyridoxal phosphate acts as an "amino carrier" - accepts the amino group → pyridoxamine phosphate → transfers to α-ketoglutarate
Key hepatic transaminases (clinical):
- ALT (Alanine aminotransferase = SGPT): Alanine + α-ketoglutarate ⇌ Pyruvate + Glutamate
- Found predominantly in liver cytosol → MOST SPECIFIC for liver injury
- AST (Aspartate aminotransferase = SGOT): Aspartate + α-ketoglutarate ⇌ OAA + Glutamate
- Found in liver, heart, muscle, RBCs → less specific
All amino acid nitrogen ultimately funnels into glutamate via transamination with α-ketoglutarate. L-glutamate is then the only amino acid that undergoes oxidative deamination at an appreciable rate.
B. Oxidative Deamination - Glutamate Dehydrogenase
L-Glutamate + NAD⁺ (or NADP⁺) → α-ketoglutarate + NH₄⁺
- Enzyme: Glutamate dehydrogenase (GDH) - located in mitochondrial matrix
- Releases free NH₄⁺ (ammonia) - the toxic product that must be detoxified
- Allosteric regulation: Activated by ADP and leucine; Inhibited by ATP and GTP (energy charge regulates NH₃ production)
- GDH links amino acid catabolism to the TCA cycle (α-ketoglutarate enters TCA)
C. Ammonia Transport to the Liver
Ammonia is highly toxic, especially to the CNS. It is transported safely from peripheral tissues to liver in two forms:
1. Glucose-Alanine Cycle (from muscle):
- Muscle: Pyruvate + Glutamate → Alanine (via ALT); Alanine exported in blood
- Liver: Alanine + α-KG → Pyruvate + Glutamate (via ALT); pyruvate → gluconeogenesis; glutamate → NH₄⁺ (via GDH) → urea cycle
2. Glutamine (from brain and other tissues):
- Glutamate + NH₄⁺ → Glutamine (enzyme: glutamine synthetase; uses ATP; occurs in brain, muscle, lung)
- Glutamine travels in blood to liver/kidney
- In liver: glutaminase → glutamate + NH₄⁺ → urea cycle
- In kidney: glutaminase → NH₄⁺ excreted in urine (important in acidosis)
5. THE UREA CYCLE (KREBS-HENSELEIT CYCLE)
The urea cycle is the primary route for disposal of ammonia. It occurs exclusively in the liver (hepatocytes). It is a bicyclic process: steps 1-2 in mitochondria; steps 3-5 in cytosol.
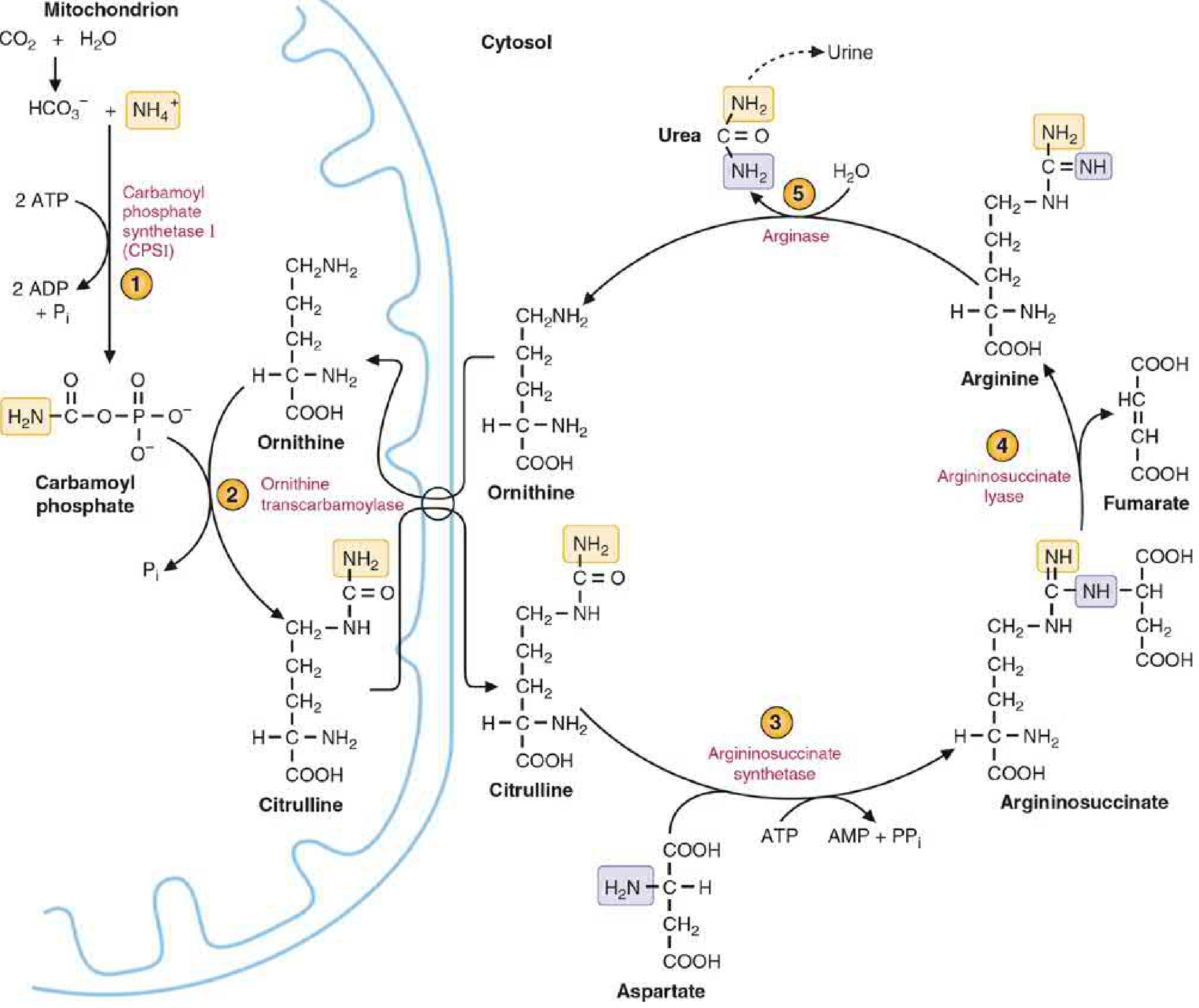
Sources of Nitrogen in Urea:
- One nitrogen from NH₄⁺ (from oxidative deamination of glutamate via GDH - mitochondrial)
- One nitrogen from aspartate (in the cytosol - donated via transamination of OAA)
- Carbon + oxygen of urea come from CO₂ (as HCO₃⁻)
- Net reaction: NH₄⁺ + HCO₃⁻ + aspartate + 3ATP → urea + fumarate + 2ADP + AMP + 4Pi
The 5 Steps:
Step 1 - Carbamoyl Phosphate Synthesis (Mitochondrial)
- NH₄⁺ + HCO₃⁻ + 2 ATP → Carbamoyl phosphate + 2 ADP + Pi
- Enzyme: Carbamoyl Phosphate Synthetase I (CPS-I)
- Location: mitochondrial matrix (of liver and intestine)
- Requires obligatory allosteric activator: N-acetylglutamate (NAG)
- Inhibited by excess NH₃ accumulation when NAG is deficient
- CPS-II (pyrimidine synthesis, cytosol, uses glutamine, NOT NAG) is a different enzyme
Step 2 - Citrulline Formation (Mitochondrial)
- Carbamoyl phosphate + Ornithine → Citrulline + Pi
- Enzyme: Ornithine Transcarbamoylase (OTC)
- X-linked; most common urea cycle defect
- Citrulline transported OUT of mitochondria (antiporter - exchanges with ornithine entering)
Step 3 - Argininosuccinate Formation (Cytosolic)
- Citrulline + Aspartate + ATP → Argininosuccinate + AMP + PPi
- Enzyme: Argininosuccinate Synthetase (ASS)
- This step incorporates the second nitrogen (from aspartate) into the cycle
Step 4 - Arginine Formation (Cytosolic)
- Argininosuccinate → Arginine + Fumarate
- Enzyme: Argininosuccinate Lyase (ASL)
- Fumarate → malate → OAA (by cytoplasmic reactions similar to TCA) → transaminated back to aspartate → re-enters cycle (this connects the urea cycle to the TCA cycle = the "argininosuccinate bridge")
Step 5 - Urea Formation and Ornithine Regeneration (Cytosolic)
- Arginine + H₂O → Urea + Ornithine
- Enzyme: Arginase I (liver cytosol); abundant in liver
- Ornithine re-enters mitochondria to restart the cycle
- Urea excreted by kidneys
Energy Cost:
- 4 high-energy phosphate bonds consumed per urea molecule: 2 ATP → 2 ADP (step 1) + 1 ATP → AMP + PPi (step 3; = 2 equivalents)
- Total = 4 "ATP equivalents" per urea synthesized
Regulation of Urea Cycle:
- Substrate availability (primary): More NH₄⁺ → more urea (feed-forward regulation). High-protein diet or starvation → urea cycle enzyme synthesis induced.
- N-Acetylglutamate (NAG): obligatory allosteric activator of CPS-I
- NAG synthesized by N-acetylglutamate synthase (NAGS): Acetyl-CoA + Glutamate → NAG
- Arginine activates NAGS → more NAG → activates CPS-I → more urea → more ornithine (via arginase) → cycle accelerates (positive feedback loop)
- Enzyme induction: High-protein diet and prolonged fasting induce all urea cycle enzyme mRNA levels (severalfold, Harper's)
Urea Cycle Disorders (Inborn Errors):
| Deficiency | Deficient Enzyme | Key Feature | Diagnostic Finding |
|---|---|---|---|
| CPS-I deficiency | CPS-I | Severe neonatal hyperammonemia | ↑NH₃, ↓citrulline, ↓arginine, normal urine orotic acid |
| OTC deficiency | Ornithine transcarbamoylase | Most common; X-linked | ↑NH₃, ↑urinary orotic acid (carbamoyl phosphate floods pyrimidine synthesis), ↓citrulline |
| Citrullinemia type I | Argininosuccinate synthetase | ↑Citrulline in blood and urine | ↑↑Citrulline, ↑NH₃ |
| Argininosuccinic aciduria | Argininosuccinate lyase | ↑Argininosuccinate; brittle hair (trichorrhexis nodosa) | ↑Argininosuccinate in urine |
| Arginase deficiency | Arginase I | ↑Arginine; progressive spastic diplegia | ↑↑Plasma arginine |
| NAGS deficiency | N-acetylglutamate synthase | Responsive to N-carbamylglutamate | ↑NH₃, normal orotic acid |
Common features of all urea cycle defects: Hyperammonemia → irritability, vomiting, lethargy → coma → death.
Treatment principles: Protein restriction + alternative waste nitrogen pathways (sodium benzoate → hippurate; sodium phenylbutyrate → phenylacetylglutamine) + arginine/citrulline supplementation.
6. BILIRUBIN METABOLISM - IN-DEPTH BIOCHEMISTRY
Step 1: Heme Catabolism → Bilirubin (in Macrophages/RES)
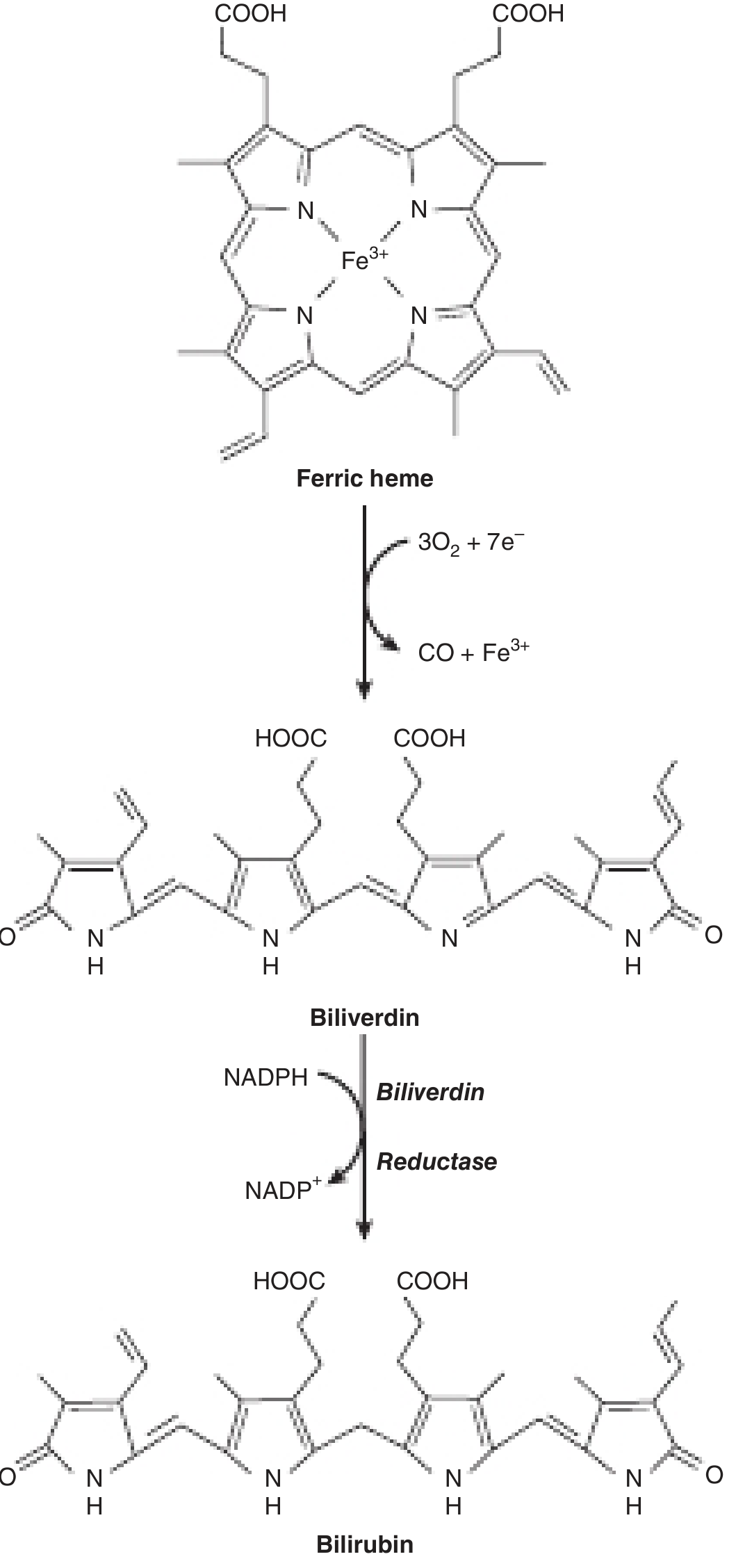
~250-350 mg bilirubin produced per day. Source breakdown:
- 65-80%: Hemoglobin breakdown (senescent RBCs, ~200 billion RBCs/day)
- 20-35%: "Early labeled peak" = ineffective erythropoiesis + myoglobin + cytochromes + peroxidases
Reaction 1 - Heme Oxygenase (microsomal):
Fe³⁺-Heme + 3O₂ + 7e⁻ (from NADPH + NADH via cytochrome P450 reductase) → Biliverdin (green) + CO + Fe³⁺
- CO produced in this reaction is endogenous CO - physiological signaling molecule; also used as biomarker of hemolysis
- Heme oxygenase is substrate-inducible (induced by its own substrate, heme)
- Inducers of heme oxygenase: hemolysis, heavy metals (Co, Cd), hypoxia
Reaction 2 - Biliverdin Reductase (cytosolic):
Biliverdin + NADPH + H⁺ → Bilirubin (yellow) + NADP⁺
- Reduces the central methylene bridge of biliverdin
- Birds and amphibians excrete biliverdin directly; humans reduce it to bilirubin
Bilirubin properties (Harper's):
- Highly lipophilic, sparingly soluble in water
- Can penetrate lipid bilayers and blood-brain barrier
- Toxic to neurons (kernicterus when unbound)
- Has internal hydrogen bonds that make it even more hydrophobic
Step 2: Transport to Liver (Albumin-Bound)
- Bilirubin + Albumin → Bilirubin-albumin complex ("indirect bilirubin")
- Albumin has a high-affinity site (binds ~25 mg bilirubin/100 mL plasma) and a low-affinity site
- Low-affinity bound bilirubin readily dissociates → tissue distribution
- Drug displacement from albumin (salicylates, sulfonamides, certain antibiotics) → releases bilirubin → risk of kernicterus in neonates
Step 3: Hepatic Uptake
- Albumin-bilirubin arrives at sinusoidal membrane (Space of Disse)
- Bilirubin dissociates from albumin
- Uptake transporters (basolateral/sinusoidal membrane):
- OATP1B1 (SLCO1B1) and OATP1B3 - sodium-independent organic anion transporters
- Some passive/facilitated diffusion also occurs
- Inside hepatocyte: bilirubin bound to Ligandin (Y protein = glutathione-S-transferase) and Z protein
- Ligandin prevents back-diffusion and intracellular sequestration in wrong compartments
- Ligandin is inducible by phenobarbital
Step 4: Conjugation with Glucuronic Acid (UDP-Glucuronosyl Transferase)
- Location: Smooth endoplasmic reticulum (microsomal fraction)
- Enzyme: UDP-glucuronosyltransferase 1A1 (UGT1A1)
- Reaction: Bilirubin + UDP-glucuronic acid → Bilirubin monoglucuronide (BG)
- Further: BG + UDP-glucuronic acid → Bilirubin diglucuronide (BG₂) [the predominant form in bile]
- A small amount is conjugated with sulfate (bilirubin sulfate) or other substances
- Conjugated bilirubin = direct bilirubin = water-soluble + non-toxic + filterable by kidney
- Phenobarbital induces UGT1A1 → used therapeutically in Crigler-Najjar type II and Gilbert's syndrome
Step 5: Canalicular Secretion
- Conjugated bilirubin secreted across apical (canalicular) membrane into bile
- Transporter: MRP2 (ABCC2 = Multidrug Resistance-associated Protein 2) - ATP-dependent active transporter
- This step is rate-limiting for overall hepatic bilirubin processing
- Defect in MRP2 → Dubin-Johnson syndrome
Step 6: Intestinal Transformation
- Bilirubin diglucuronide → terminal ileum/colon → bacterial beta-glucuronidases → deconjugated → free bilirubin → further bacterial reduction → urobilinogen (colorless) → urobilin (yellow, oxidized in urine) or stercobilin (brown, in feces)
Van den Bergh Reaction (Clinical Lab Test):
| Form | Reaction with diazo reagent | Name | Solubility |
|---|---|---|---|
| Unconjugated bilirubin | Reacts ONLY after adding ethanol/methanol (alcohol) | "Indirect" | Lipid-soluble, insoluble in water |
| Conjugated bilirubin | Reacts DIRECTLY (no alcohol needed) | "Direct" | Water-soluble |
| Total bilirubin | After adding alcohol | Indirect + Direct | - |
Normal values:
- Total bilirubin: 0.2 - 1.0 mg/dL
- Direct (conjugated): 0.0 - 0.3 mg/dL
- Indirect (unconjugated): 0.2 - 0.8 mg/dL
Genetic Disorders of Bilirubin Metabolism (Harper's)
| Condition | Enzyme/Protein Defect | Bilirubin type | Clinical | Treatment |
|---|---|---|---|---|
| Neonatal physiologic jaundice | Immature UGT1A1 + hemolysis | Unconjugated | Transient; if severe → kernicterus | Phototherapy (isomerizes bilirubin to water-soluble isoforms excreted in bile/urine) |
| Gilbert syndrome | UGT1A1 activity ~30% of normal (TATA box promoter mutation, A(TA)7TAA) | Unconjugated (mild, <3 mg/dL) | Benign; jaundice with fasting, illness, stress; no treatment needed | None required |
| Crigler-Najjar type I | Complete UGT1A1 absence | Unconjugated (>20 mg/dL) | Severe kernicterus; fatal in infancy without treatment | Daily 10-12 hr phototherapy; liver transplant (curative) |
| Crigler-Najjar type II | Partial UGT1A1 (some activity retained) | Unconjugated (<20 mg/dL) | Less severe; responds to phenobarbital | Phenobarbital (induces residual UGT1A1) |
| Dubin-Johnson syndrome | MRP2 (ABCC2) defect - canalicular secretion | Conjugated | Benign; black/dark liver (melanin-like pigment accumulation); ↑urinary coproporphyrin I (>80%) | None |
| Rotor syndrome | OATP1B1 + OATP1B3 deficiency (impaired hepatic uptake and storage) | Conjugated | Benign; NO liver pigment; ↑urinary coproporphyrin I + III | None |
7. HEPATIC DRUG METABOLISM AND DETOXIFICATION
The liver is the primary xenobiotic-metabolizing organ. The goal is to convert lipophilic compounds (which would accumulate in tissues) to hydrophilic metabolites for urinary or biliary excretion.
Phase I Reactions (Functionalization)
- Oxidation, reduction, hydrolysis - introduce or expose a functional group (–OH, –NH₂, –COOH, –SH)
- Primarily by Cytochrome P450 (CYP) enzymes - collectively called "Mixed Function Oxidases (MFO)" or "Microsomal Ethanol Oxidizing System (MEOS)"
- Location: Smooth endoplasmic reticulum (microsomal fraction)
- Mechanism: CYP + NADPH + O₂ → metabolite + NADP⁺ + H₂O; generates a reactive free radical intermediate
Key CYP isoforms:
| CYP Isoform | Substrates | Clinical notes |
|---|---|---|
| CYP3A4 | Majority of drugs (>50%); statins, cyclosporine, HIV protease inhibitors, benzodiazepines | Induced by rifampin, St John's Wort; inhibited by ketoconazole, grapefruit juice |
| CYP2D6 | Codeine, beta-blockers, antidepressants | Genetic polymorphism - poor vs. extensive metabolizers |
| CYP2C9 | Warfarin (S-warfarin), NSAIDs, phenytoin | Bleeding risk with inhibitors |
| CYP2C19 | Omeprazole, clopidogrel (activation), diazepam | |
| CYP2E1 | Ethanol, acetaminophen, isoflurane, carbon tetrachloride | Induced by ethanol; generates NAPQI from acetaminophen |
| CYP1A2 | Caffeine, theophylline, certain carcinogens |
Properties of all CYP enzymes (Basic Med Biochemistry):
- All are found in smooth ER (microsomal fraction)
- All bound to phospholipid (phosphatidylcholine) in membrane
- All are inducible by their own substrates (and less so by other substrates)
- All generate a reactive free-radical intermediate
Clinical: Acetaminophen (Paracetamol) Hepatotoxicity - The Classic Example (Basic Med Biochemistry):
At therapeutic doses:
- Acetaminophen → glucuronide conjugate (by UGT, ~55%) + sulfate conjugate (by SULT, ~30%) → safe renal excretion
- Small amount (~5-10%) → CYP2E1 → NAPQI (N-acetyl-p-benzoquinoneimine) → conjugated with glutathione → safe mercapturic acid → excreted
In overdose (or in alcoholics where CYP2E1 is induced):
- Glucuronide and sulfate pathways saturated
- NAPQI production overwhelms glutathione stores
- Free NAPQI → covalently binds liver cell proteins → centrilobular (zone 3) necrosis
- Treatment: N-acetylcysteine (NAC) replenishes glutathione stores
Aflatoxin B1 Bioactivation:
- Aflatoxin B1 (from Aspergillus flavus on peanuts/stored grains) → CYP2A1 → Aflatoxin B1-8,9-epoxide
- Epoxide forms covalent adducts with guanine in DNA → G→T transversion in p53 tumor suppressor gene (codon 249) → hepatocellular carcinoma (HCC)
- Major cause of HCC in sub-Saharan Africa and Southeast Asia
Phase II Reactions (Conjugation/Biotransformation)
Make metabolites more polar/water-soluble by conjugation:
| Reaction | Conjugating group | Enzyme | Product |
|---|---|---|---|
| Glucuronidation | Glucuronic acid (from UDP-glucuronate) | UDP-glucuronosyltransferase (UGT) | Glucuronide conjugate (excreted in bile or urine) |
| Sulfation | Sulfate (from PAPS = 3'-phosphoadenosine-5'-phosphosulfate) | Sulfotransferase (SULT) | Sulfate ester |
| Glutathione conjugation | Glutathione (GSH) | Glutathione-S-transferase (GST) | Mercapturic acid (after further processing) |
| Acetylation | Acetyl group (from acetyl-CoA) | N-acetyltransferase (NAT) | N-acetyl derivative |
| Methylation | Methyl group (from SAM - S-adenosylmethionine) | Methyltransferase | Methylated compound |
| Amino acid conjugation | Glycine, glutamine | Acyl-CoA:amino acid transferase | Hippuric acid (benzoate + glycine), phenylacetylglutamine |
8. PLASMA PROTEIN SYNTHESIS BY THE LIVER
The liver synthesizes virtually all plasma proteins except immunoglobulins (made by plasma cells) and von Willebrand factor (endothelium).
Albumin (Most Important - Basic Medical Biochemistry)
- MW = 69 kDa (smallest major plasma protein)
- Normal serum concentration: 3.5-5.0 g/dL
- Constitutes ~60% of total plasma protein but contributes 70-80% of plasma oncotic pressure (due to small size and high concentration - Starling forces)
- Synthesized exclusively by hepatocytes (~12 g/day in adults)
- Half-life: 20 days → falls slowly in chronic liver disease (not acute)
- Functions as carrier protein for:
- Free fatty acids (FFAs)
- Bilirubin (unconjugated)
- Calcium (40% of plasma Ca is albumin-bound)
- Zinc, copper
- Steroid hormones (cortisol, estrogen, testosterone)
- Thyroid hormone (T4)
- Many drugs (warfarin, NSAIDs, digoxin, benzodiazepines)
- Negative acute phase protein - albumin synthesis decreases during inflammation (shift to acute phase protein production)
Binding Proteins Synthesized by Liver (Basic Med Biochemistry Table)
| Protein | Binds/Transports |
|---|---|
| Albumin | FFAs, bilirubin, Ca²⁺, drugs, steroids, thyroid hormone |
| Transferrin | Iron (Fe³⁺); saturated ~30% normally |
| Ferritin | Intracellular iron storage (not a plasma transport protein) |
| Ceruloplasmin | Copper transport; ferroxidase activity; ↓in Wilson's disease |
| Haptoglobin | Free hemoglobin (prevents iron loss; ↓in hemolysis) |
| Retinol-binding protein | Vitamin A (retinol) |
| Transthyretin (Prealbumin) | Thyroxine (T4) + forms complex with RBP |
| Sex hormone-binding globulin (SHBG) | Estradiol + testosterone |
| Corticosteroid-binding globulin (Transcortin, CBG) | Cortisol |
| Lipoproteins | Cholesterol, fatty acids (VLDL, HDL) |
| Thyroid binding globulin (TBG) | T4, T3 |
| Alpha-1-antitrypsin (α1-AT) | Serine protease inhibitor (elastase, trypsin); deficiency → emphysema + liver disease |
Acute Phase Proteins (APPs)
During acute infection/inflammation, IL-1, IL-6, TNF-α from macrophages signal liver to alter protein synthesis:
Positive APPs (↑ during inflammation) - synthesized more:
- C-Reactive Protein (CRP) - 1000-fold increase; activates complement; opsonin
- Serum amyloid A (SAA)
- Fibrinogen (↑ clotting risk)
- Alpha-1-antitrypsin
- Haptoglobin
- Ceruloplasmin
- Complement C3, C4
- Ferritin
Negative APPs (↓ during inflammation) - synthesized less:
- Albumin (major - explains low albumin in chronic inflammation and sepsis)
- Transferrin
- Transthyretin (prealbumin)
- RBP (retinol-binding protein)
Clotting Factors Synthesized by Liver
| Factor | Name | Vitamin K-dependent? |
|---|---|---|
| Factor I | Fibrinogen | No |
| Factor II | Prothrombin | Yes |
| Factor V | Labile factor | No |
| Factor VII | Proconvertin | Yes |
| Factor IX | Christmas factor | Yes |
| Factor X | Stuart-Prower factor | Yes |
| Factor XI | Plasma thromboplastin antecedent | No |
| Protein C | Anticoagulant | Yes |
| Protein S | Anticoagulant (cofactor for Protein C) | Yes |
| Antithrombin | Serine protease inhibitor | No |
Vitamin K-dependent factors (II, VII, IX, X + Protein C + Protein S) require carboxylation of glutamic acid residues (γ-carboxylation) by vitamin K-dependent gamma-carboxylase → allows calcium binding and phospholipid membrane attachment needed for clotting cascade.
Warfarin inhibits Vitamin K epoxide reductase (VKOR) → prevents recycling of Vitamin K → reduced synthesis of these factors → anticoagulation.
In liver disease:
- PT/INR rises early (Factor VII has shortest half-life ~4-6 hours) → best test of acute hepatic synthetic function
- In chronic liver disease: all factors fall → PT/INR ↑ + PTT ↑
- Factor V is synthesized by liver but is NOT Vitamin K-dependent → Factor V level can distinguish hepatocellular failure from Vitamin K deficiency/warfarin effect
9. HEPATIC METABOLISM OF ETHANOL
Ethanol metabolism in the liver is central to alcoholic liver disease:
Three Pathways:
1. Alcohol Dehydrogenase (ADH) Pathway - Main route (low alcohol concentrations):
- Ethanol + NAD⁺ → Acetaldehyde + NADH (ADH; cytosol)
- Acetaldehyde + NAD⁺ → Acetate + NADH (ALDH2 = aldehyde dehydrogenase 2; mitochondria)
- Acetate → blood → peripheral tissues → acetyl-CoA
2. Microsomal Ethanol Oxidizing System (MEOS) - CYP2E1 Pathway (high alcohol concentrations, induced by chronic use):
- Ethanol + NADPH + H⁺ + O₂ → Acetaldehyde + NADP⁺ + 2H₂O
- CYP2E1 is inducible (explains tolerance in alcoholics and increased NAPQI generation)
- Generates reactive oxygen species (ROS) → oxidative stress → lipid peroxidation
3. Catalase Pathway (minor):
- Ethanol + H₂O₂ → Acetaldehyde + 2H₂O (requires H₂O₂ generated by other reactions)
Metabolic Consequences of Excess NADH Production:
The conversion of ethanol to acetaldehyde and acetate generates massive amounts of NADH in the liver. The resulting elevated NADH:NAD⁺ ratio has multiple biochemical consequences:
| Consequence | Mechanism |
|---|---|
| Hypoglycemia | ↑NADH → ↓NAD⁺ → inhibits gluconeogenesis (NAD⁺ required for glycerol-3-phosphate and lactate oxidation steps); also inhibits pyruvate carboxylase |
| Hyperlactatemia / Lactic acidosis | ↑NADH favors lactate over pyruvate (LDH equilibrium shifts); reduces lactate → pyruvate for gluconeogenesis |
| Hyperlipidemia / Fatty liver | ↑NADH inhibits beta-oxidation (NAD⁺-dependent steps in beta-oxidation are reversed); excess acetyl-CoA → fatty acid synthesis → hepatic fat accumulation → steatosis |
| Hyperuricemia | Lactate competes with uric acid for renal tubular secretion |
| Inhibition of TCA cycle | High NADH inhibits isocitrate dehydrogenase and alpha-ketoglutarate dehydrogenase |
| Ketogenesis | Excess acetyl-CoA → ketone body synthesis |
Acetaldehyde toxicity:
- Highly reactive; forms covalent adducts with proteins → impairs protein function
- Binds tubulin → impairs hepatocyte secretion (fat + protein accumulate in cells)
- Binds mitochondrial proteins → mitochondrial dysfunction
- Stimulates stellate cell activation → collagen synthesis → fibrosis
10. LIVER FUNCTION TESTS - BIOCHEMICAL BASIS (Basic Medical Biochemistry)
Enzymes (Reflect Hepatocyte Injury):
| Test | What It Is | Pattern |
|---|---|---|
| ALT (Alanine Aminotransferase = SGPT) | Cytosolic enzyme; most abundant in hepatocytes; requires PLP (B₆) | Most specific for hepatocyte necrosis; 10-100x rise in viral hepatitis, drug-induced hepatitis; milder in alcoholic hepatitis (ALT/AST < 1) |
| AST (Aspartate Aminotransferase = SGOT) | Cytosolic + mitochondrial isoforms; in liver, heart, muscle, brain, RBCs; requires PLP (B₆) | Elevated in hepatitis + MI + muscle disease; AST:ALT > 2:1 = alcoholic hepatitis (alcohol damages mitochondria → mitochondrial AST released) |
| ALP (Alkaline Phosphatase) | Canalicular membrane enzyme; in liver, bone, intestine, placenta | Elevated in cholestasis (intrahepatic or extrahepatic obstruction) and bone disease |
| GGT (Gamma-Glutamyl Transpeptidase) | Cholangiocyte membrane; catalyzes glutathione hydrolysis | ↑ in cholestasis + alcohol abuse; confirms ALP elevation is hepatic (not bone); highly inducible by alcohol + drugs |
| 5'-Nucleotidase (5'-NT) | Sinusoidal membrane enzyme; liver + intestine only | Elevated in cholestasis; more specific than ALP for liver (not elevated in bone disease) |
Synthetic Function Tests:
| Test | Biochemical Basis | Half-life | Reflects |
|---|---|---|---|
| Serum Albumin | Synthesized by liver only | 20 days | Chronic hepatic synthetic function; falls in chronic liver disease, cirrhosis, malnutrition, protein-losing enteropathy |
| Prothrombin Time (PT) / INR | Factors II, V, VII, X (all liver-synthesized; II, VII, X are Vit K-dependent); Factor VII has shortest half-life | Factor VII = 4-6 hrs | Acute hepatic synthetic function; rises within hours-days in acute liver failure |
| Serum globulins | Elevated globulins (IgG, IgA, IgM) in liver disease | - | Reflect immune activation; polyclonal hypergammaglobulinemia common in cirrhosis |
Bilirubin Tests:
| Test | Interpretation |
|---|---|
| Total bilirubin | >1.5 mg/dL = clinical jaundice; >2.5-3 mg/dL = visible jaundice |
| Direct bilirubin | Conjugated; elevated in cholestasis and hepatocellular disease |
| Indirect bilirubin | Unconjugated; elevated in hemolysis (pre-hepatic) and Gilbert's/Crigler-Najjar (hepatic conjugation failure) |
| Urine bilirubin | Only conjugated bilirubin (water-soluble) appears in urine; absent in pre-hepatic jaundice |
| Urine urobilinogen | Elevated in hemolysis and hepatocellular disease; absent in complete biliary obstruction |
Hepatic Injury Patterns (Basic Medical Biochemistry):
| Pattern | Main abnormality | Causes |
|---|---|---|
| Hepatocellular | ↑↑ALT + ↑AST > ↑ALP | Viral hepatitis, drug-induced hepatitis, autoimmune hepatitis |
| Cholestatic | ↑↑ALP + ↑GGT > ↑ALT | Biliary obstruction (stones, cancer), PBC, PSC, drugs |
| Mixed | Both ALT and ALP elevated | Some drug reactions, overlap syndromes |
| Infiltrative | ↑ALP + normal or mildly ↑ALT | Metastases, granulomas, amyloidosis |
11. ONE-CARBON METABOLISM AND THE LIVER
The liver is central to one-carbon transfer reactions using folate and B₁₂:
S-Adenosylmethionine (SAM) - Universal Methyl Donor
- Methionine + ATP → SAM (by methionine adenosyltransferase; abundant in liver)
- SAM donates methyl groups to: DNA (methylation of cytosine), RNA, proteins, phospholipids (PC synthesis from PE), norepinephrine → epinephrine, guanidinoacetate → creatine, arsenic detoxification
- After donating methyl group: SAM → S-adenosylhomocysteine (SAH) → homocysteine
- Homocysteine remethylation: Homocysteine + 5-methylTHF → methionine + THF (requires Vitamin B₁₂ as cofactor for methionine synthase)
- Homocysteine transsulfuration: Homocysteine + serine → cystathionine → cysteine (requires Vitamin B₆ as cofactor)
- In liver disease: impaired SAM synthesis → impaired methylation → affects phospholipid synthesis, DNA methylation
Clinical: Homocysteinemia
- Elevated homocysteine → endothelial damage → atherosclerosis risk
- Caused by: Vitamin B₁₂, B₆, or folate deficiency; cystathionine beta-synthase deficiency (homocystinuria)
- Liver disease impairs folate/B₁₂ metabolism → contributes to elevated homocysteine
12. GLYCOGEN STORAGE DISEASES - HEPATIC FORMS
| Disease | Deficient Enzyme | Liver findings | Clinical |
|---|---|---|---|
| Type I - Von Gierke | Glucose-6-phosphatase | Massive hepatomegaly; ↑glycogen + fat | Severe fasting hypoglycemia, lactic acidosis, hyperlipidemia, hyperuricemia; doll-like face |
| Type III - Cori/Forbes | Debranching enzyme | Hepatomegaly | Milder hypoglycemia than Type I; ↑glycogen with short outer chains |
| Type IV - Andersen | Branching enzyme | Cirrhosis, liver failure | Progressive liver disease; storage of abnormal glycogen (amylopectin-like) |
| Type VI - Hers | Hepatic glycogen phosphorylase | Hepatomegaly | Mild hypoglycemia |
| Type VIII/IX | Phosphorylase kinase | Hepatomegaly | Mild; usually benign |
Sources: Harper's Illustrated Biochemistry, 32nd Ed (9781260469943), Lippincott Illustrated Reviews: Biochemistry, 8th Ed (9781975155063), Basic Medical Biochemistry: A Clinical Approach, 6th Ed (9781975150143)
That completes the full biochemistry of the liver - covering carbohydrate metabolism (glycolysis, glycogenesis, glycogenolysis, gluconeogenesis), lipid metabolism (beta-oxidation, ketogenesis, lipogenesis, cholesterol synthesis, bile acid synthesis, lipoproteins), protein/amino acid metabolism (transamination, urea cycle with all 5 steps and defects), bilirubin metabolism at the molecular level, detoxification/CYP450 system, plasma protein synthesis, ethanol metabolism, and LFT biochemical interpretation.
Next topic:
- Pathology (Robbins - hepatitis, cirrhosis, NAFLD, HCC histopathology)
- Microbiology (Hepatitis A-E viruses)
- Pharmacology (drugs for liver disease)
- Medicine (clinical management - Harrison's)
- Surgery (hepatic resection, portal hypertension surgery)
Patho
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is the complete textbook-level pathology of the liver, synthesized from Robbins & Cotran Pathologic Basis of Disease (full edition) and Robbins & Kumar Basic Pathology.
PATHOLOGY OF THE LIVER - Full Textbook-Level Notes (Robbins)
1. NORMAL LIVER HISTOLOGY - PATHOLOGICAL BASELINE
The Lobular Model (used by pathologists):
- Terminal hepatic vein (central hepatic vein) = center
- Portal tracts at periphery
- Periportal = near portal tract; Centrilobular (perivenular) = near central vein
- Blood flows: portal tract sinusoids → central vein
The Acinar Model (functional):
- Zone 1 (periportal) = closest to blood supply → best oxygenated → zone of gluconeogenesis, beta-oxidation, bile acid synthesis
- Zone 2 = intermediate
- Zone 3 (centrilobular/perivenular) = farthest from blood supply → lowest O₂ → zone of glycolysis, lipid synthesis, drug metabolism (CYP450) → most susceptible to ischemic/toxic injury, alcohol, acetaminophen
Key histological cell types:
- Hepatocytes - polyhedral cells with round nuclei, prominent nucleoli; arranged in 1-2 cell thick plates
- Kupffer cells - liver macrophages on luminal face of sinusoids
- Stellate cells (Ito/lipocyte cells) - in Space of Disse; store Vitamin A; when activated → myofibroblasts → fibrosis
- Sinusoidal endothelium - fenestrated (no basement membrane); overlies Space of Disse
The portal triad contains: portal vein branch + hepatic artery branch + bile duct
Bile canaliculi = channels 1-2 µm formed by grooves between adjacent hepatocytes → drain into canals of Hering → bile ductules → terminal bile ducts in portal tracts
2. HEPATIC FAILURE
Acute Liver Failure (Basic Pathology)
- Defined as acute liver illness producing hepatic encephalopathy within 6 months of initial diagnosis
- USA causes: Acetaminophen (paracetamol) overdose ~50% of ALF; autoimmune hepatitis, other drugs/toxins, acute hepatitis A and B
- Asia: acute hepatitis B and E predominate
Morphology - Massive Hepatic Necrosis:
- Gross: Small, shrunken liver (may weigh as little as 700 g); bile-stained; soft, congested
- Micro: Large zones of destruction (zonal necrosis - zone 3 in acetaminophen, zone 1 in phosphorus); occasional islands of regenerating hepatocytes; minimal scarring (acute process, no time to fibrosis)
Clinical Features of ALF:
- Jaundice, nausea, vomiting, fatigue
- Hepatic encephalopathy (most ominous sign) - due to ↑NH₃ (urea cycle fails) + GABA accumulation + false neurotransmitters + short-chain fatty acids → cerebral edema
- Coagulopathy (↑PT/INR - clotting factors fall rapidly)
- Portal hypertension → ascites
- Serum transaminases initially very high → then fall sharply as hepatocytes disappear (not a good sign - "burnout")
- Eventual multiorgan failure → death without liver transplantation
Complications:
- Hepatorenal syndrome (HRS): Functional renal failure without intrinsic renal disease; caused by splanchnic vasodilation → ↑renin-angiotensin → severe renal vasoconstriction → ↓GFR; reversible with liver transplant
- Coagulopathy: Combined - ↓clotting factors (liver synthesis fails) + ↑consumption (DIC tendency)
- Hepatic encephalopathy: Asterixis (flapping tremor), confusion, coma
3. CIRRHOSIS
Definition and General Features (Robbins & Cotran)
Cirrhosis = diffuse remodeling of the entire liver into parenchymal nodules surrounded by fibrous bands. It is a whole-organ diagnosis - not focal.
Key elements:
- Parenchymal nodules (usually regenerative)
- Dense fibrous bands surrounding nodules - portal-to-portal bridging, or portal-to-central bridging
- Altered vascular architecture - portosystemic shunting, loss of sinusoidal organization
- Variable degree of ongoing injury, repair, regeneration
Leading causes worldwide:
- Chronic Hepatitis B
- Chronic Hepatitis C
- MASLD (Metabolic dysfunction-Associated Steatotic Liver Disease = NAFLD)
- Alcoholic-associated liver disease
- Cryptogenic (no identified cause)
Morphology:
Gross:
- Bumpy surface (depressed scarring alternating with bulging regenerative nodules)
- Normal smooth capsule replaced by irregular surface
- Liver initially enlarged (fatty/viral) → then shrunken (end-stage)
Microscopic:
- Fibrous bands: highlighted by special stains (Masson's trichrome = blue collagen; Sirius Red = red collagen)
- Nodule classification:
- Micronodular (<3 mm): uniform small nodules - seen in alcoholic cirrhosis, hemochromatosis, biliary cirrhosis
- Macronodular (>3 mm, variable size): seen in chronic viral hepatitis, autoimmune hepatitis (regeneration after subacute necrosis)
- Mixed: both patterns coexist
- Features of regression: thin, incomplete scars; variable ductular reaction
Morphologic note: Cirrhosis is not synonymous with end-stage liver disease. Not all cirrhosis leads to failure (well-treated AIH, cured HCV may not progress).
Pathogenesis of Fibrosis and Cirrhosis
The common pathway for all forms of cirrhosis is hepatic stellate cell (HSC) activation:
- Liver injury (alcohol, virus, toxin, metabolic) → hepatocyte death → inflammatory cell recruitment
- Kupffer cell activation → releases: TGF-β1 (most potent fibrogenic cytokine), TNF-α, IL-1, ROS, platelet-derived growth factor (PDGF)
- Hepatic stellate cells (Ito cells) in Space of Disse:
- Normally store vitamin A (quiescent phenotype)
- Activated by: TGF-β1, PDGF, ROS, acetaldehyde (in alcohol)
- Become myofibroblasts → proliferate, lose vitamin A, migrate, contract sinusoids
- Secrete type I and III collagen (fibrosis) and MMP inhibitors (TIMP-1, TIMP-2) → prevents collagen breakdown
- Fibrous bands form → distort lobular architecture → portosystemic shunts → portal hypertension
- Cirrhosis is potentially reversible in early stages if injury removed (HCV cure, alcohol cessation) → HSCs undergo apoptosis, MMPs degrade collagen → regression of fibrosis is documented
4. PORTAL HYPERTENSION
Causes (Robbins & Cotran Table 18.2)
| Location | Causes |
|---|---|
| Pre-hepatic | Portal vein thrombosis; narrowing of portal vein; massive splenomegaly (↑splenic venous flow) |
| Intrahepatic (most common = cirrhosis) | Cirrhosis (any cause); nodular regenerative hyperplasia; PBC; schistosomiasis; massive fatty change; granulomatous disease (sarcoid); infiltrative malignancy; amyloidosis |
| Post-hepatic | Severe right heart failure; constrictive pericarditis; hepatic vein outflow obstruction (Budd-Chiari) |
Pathophysiology (Robbins & Cotran)
Two components:
- ↑ Resistance to portal flow at sinusoidal level:
- Myofibroblast/vascular smooth muscle contraction
- Scarring and nodule formation
- ↓ NO production by sinusoidal endothelium + ↑ endothelin-1 + ↑ angiotensinogen → intrahepatic vasoconstriction
- Sinusoidal arterio-portal shunts (arterial pressure transmitted to portal system)
- ↑ Portal venous blood flow via hyperdynamic splanchnic circulation:
- Splanchnic arterial vasodilation (mediated primarily by NO, also prostacyclin, TNF-α) → ↑ splanchnic blood flow → ↑ portal venous inflow
Four Major Consequences of Portal Hypertension:
1. Ascites:
- Accumulation of fluid in the peritoneal cavity
- Mechanism: portal hypertension → hydrostatic pressure ↑ + hypoalbuminemia → ↑ sinusoidal ultrafiltration; splanchnic vasodilation → ↓ effective circulating volume → ↑ RAA system activation → Na⁺ and water retention
- High protein ascites (exudate-like): in early liver disease (sinusoids very leaky)
- Low protein ascites (transudate-like): in later disease with hypoalbuminemia
- SAAG (serum-ascites albumin gradient) > 1.1 g/dL indicates portal hypertension
2. Portosystemic Venous Shunts:
- As portal pressure rises → portal blood diverted through collateral channels to systemic veins
- Esophageal varices: Left gastric (coronary) vein → esophageal veins → azygos → most dangerous - rupture → massive hematemesis (mortality 20-30% per episode)
- Caput medusae: Paraumbilical veins → superficial abdominal wall veins → radiating from umbilicus
- Anorectal varices (hemorrhoids): Superior rectal (portal) → middle/inferior rectal (systemic)
- Retroperitoneal shunts (Retzius veins): Usually asymptomatic
3. Congestive Splenomegaly:
- Portal hypertension → splenic venous congestion → splenomegaly
- Hypersplenism → ↑ destruction of blood cells → thrombocytopenia (most common, first) + anemia + leukopenia
- Platelet count typically 50,000-100,000/µL in moderate hypersplenism
4. Hepatic Encephalopathy (see Liver Failure section)
Hepatorenal Syndrome:
- As discussed - functional renal failure; renin-angiotensin activation worsens renal perfusion; principally in cirrhosis, severe alcoholic hepatitis, fulminant failure
5. VIRAL HEPATITIS
Clinicopathologic Syndromes (Common to All Hepatotropic Viruses)
A. Acute Hepatitis - Morphology
-
Gross: Enlarged, soft, red-brown congested liver
-
Micro:
- Hepatocyte injury patterns: Ballooning degeneration (swollen, clear cytoplasm - earliest sign), eosinophilic degeneration
- Apoptosis: Acidophil bodies (Councilman bodies) = eosinophilic, rounded, shrunken hepatocytes with pyknotic nuclei - marker of hepatocyte apoptosis
- Lobular disarray: Loss of normal plate architecture
- Inflammation: Mixed portal and intralobular infiltrate of lymphocytes, macrophages; neutrophils in alcoholic hepatitis
- Cholestasis: Bile pigment in hepatocytes and canaliculi
- Kupffer cell hyperplasia: Enlarged, pigment-laden Kupffer cells
- Fatty change (steatosis): Particularly in HCV
-
Severe acute hepatitis: Bridging necrosis = confluent necrosis connecting portal tracts to central veins (or portal-to-portal or central-to-central) - indicates bad prognosis
B. Chronic Hepatitis - Morphology (>6 months)
Key histological features:
- Portal inflammation: Lymphocytic infiltrate in portal tracts (± germinal centers in HCV)
- Interface hepatitis (Piecemeal necrosis) = lymphocyte-mediated destruction of hepatocytes at the limiting plate (junction of portal tract and parenchyma) = cardinal feature of chronic hepatitis
- Lobular activity: Scattered hepatocyte necrosis and inflammation throughout lobule
- Fibrosis: Portal → periportal → bridging → cirrhosis (Metavir scoring F0-F4)
- Mallory-Denk bodies: Eosinophilic hyaline inclusions of cytokeratin intermediate filaments in hepatocytes - seen in alcoholic hepatitis but also in NASH
- Grading (activity = degree of inflammation) and Staging (fibrosis progression)
| Hepatitis Virus | Specific Histological Features |
|---|---|
| HBV | Ground-glass hepatocytes (HBsAg accumulation in ER); "sanded nuclei" (HBcAg in nucleus); minimal steatosis |
| HCV | Lymphoid follicles in portal tracts; bile duct damage (lymphocytes invading bile duct epithelium); steatosis (macrovesicular) particularly in genotype 3 |
| Autoimmune | Abundant plasma cells; emperipolesis (lymphocytes within hepatocyte cytoplasm); rosettes |
Hepatitis A Virus (HAV)
| Feature | Details |
|---|---|
| Virus | Picornavirus (Hepatovirus genus); small, non-enveloped, positive-sense ssRNA; 27 nm, icosahedral |
| Receptor | HAVcr-1 (TIM-1) on hepatocytes |
| Transmission | Fecal-oral (contaminated water, food, shellfish); NO blood transmission (viremia transient) |
| Incubation | 2-6 weeks |
| Pathogenesis | NOT cytopathic directly; injury by cytotoxic T lymphocytes and NK cells |
| Chronicity | NEVER - always self-limited |
| Diagnosis | Serum IgM anti-HAV (acute); IgG anti-HAV (past/immunity) |
| Acute liver failure | 0.1-0.3% (higher in those with pre-existing liver disease) |
| Carrier state | None |
| Prevention | Inactivated HAV vaccine (excellent efficacy) |
Hepatitis B Virus (HBV)
| Feature | Details |
|---|---|
| Virus | Hepadnavirus; partially double-stranded DNA; 42 nm Dane particle (complete virus) |
| Transmission | Parenteral (IV drug use, needlestick, transfusion); Sexual; Perinatal (vertical - main route in Asia) |
| Incubation | 2-26 weeks |
| Chronicity | 5-10% of adults; 90% in neonates (immature immune response) |
HBV Antigens and Antibodies - Complete Serological Profile:
| Marker | Produced by | Significance |
|---|---|---|
| HBsAg (Surface Antigen) | Envelope protein; excess secreted into blood | First marker to appear in acute infection; present in carrier state; its persistence >6 months = chronic HBV |
| Anti-HBs | Antibody to HBsAg | Appears after recovery or vaccination; indicates immunity |
| HBcAg (Core Antigen) | Core protein | NOT detected in serum (only in liver cells); detected by immunostaining in biopsy (cytoplasmic = active replication; nuclear = low replication) |
| Anti-HBc IgM | Antibody to core antigen | Appears early in acute infection; ONLY marker present during the "window period" (after HBsAg falls, before anti-HBs appears) |
| Anti-HBc IgG | Indicates past infection; persists lifelong | |
| HBeAg (e Antigen) | Secreted form of core protein | Marker of active viral replication and high infectivity; correlates with HBV DNA levels |
| Anti-HBe | Antibody to HBeAg | Seroconversion indicates ↓ replication; lower infectivity; may indicate "pre-core mutant" HBV |
| HBV DNA | Viral genome | Best direct measure of viral replication (quantitative PCR) |
Window Period: Time between disappearance of HBsAg and appearance of Anti-HBs. During this period, Anti-HBc IgM is the only diagnostic marker.
HBV Morphology:
- Ground-glass hepatocytes: Finely granular, eosinophilic cytoplasm due to massive HBsAg accumulation in ER (PAS negative, Orcein positive, Victoria blue stain positive)
- Sanded nuclei: HBcAg in nuclei creates ground-glass nuclear appearance
- Minimal steatosis (unlike HCV)
Outcome of HBV:
- Acute symptomatic infection → self-limited recovery (95% of adults)
- Chronic HBV → cirrhosis → HCC (risk 200x higher than uninfected)
- Precore mutant HBV: HBeAg negative but HBV DNA positive (mutant virus cannot produce HBeAg)
Hepatitis C Virus (HCV)
| Feature | Details |
|---|---|
| Virus | Flavivirus family; positive-sense ssRNA; envelope glycoproteins E1, E2 (highly variable) |
| Transmission | Parenteral (IVDU main route); intranasal cocaine; sexual (low risk); perinatal (low risk) |
| Incubation | 4-26 weeks |
| Chronicity | >80% → most common cause of chronic hepatitis in developed world |
HCV Morphology (Distinctive Features):
- Lymphoid follicles in portal tracts (germinal centers = hallmark)
- Bile duct damage - lymphocytes infiltrating bile duct epithelium (mimics PBC)
- Macrovesicular steatosis - especially genotype 3 (direct viral effect on lipid metabolism)
- Interface hepatitis with lobular activity
- Progression: fibrosis in 20-30 years → cirrhosis → HCC
Diagnosis: ELISA for anti-HCV antibodies (screening); quantitative HCV RNA PCR (confirmation, monitoring treatment)
- Anti-HCV does NOT distinguish acute from chronic or indicate immunity
- False positive anti-HCV: low albumin states (EIA can give false positives)
HCV Serology:
- No vaccine available (high genetic variability, quasispecies, immune escape)
- Treatment: Direct-acting antivirals (DAAs) → >95% cure rate (SVR = sustained virological response)
Hepatitis D Virus (HDV)
| Feature | Details |
|---|---|
| Virus | Defective circular ssRNA (delta agent) - requires HBsAg as its envelope (obligate parasite of HBV) |
| Transmission | Parenteral (same as HBV) |
| Forms of infection | Co-infection (simultaneous HBV + HDV): severe acute hepatitis but usually resolves; Superinfection (HDV in chronic HBV carrier): severe, high risk of fulminant hepatitis and chronic HDV |
| Chronicity | Co-infection: 5-10%; Superinfection: 90-100% |
| Prognosis | HDV superinfection has worst prognosis of all hepatotropic viruses |
Hepatitis E Virus (HEV)
| Feature | Details |
|---|---|
| Virus | Non-enveloped positive-sense ssRNA; Hepeviridae family |
| Transmission | Fecal-oral (waterborne; undercooked pork/game meat in high-income countries) |
| Incubation | 4-5 weeks |
| Chronicity | Only in immunocompromised hosts (transplant recipients); never in immunocompetent |
| Special risk | Fulminant hepatitis in pregnancy (especially 3rd trimester) - mortality up to 20-25% |
| Geography | Endemic in Asia, Africa, Mexico, Central America; sporadic in developed countries (HEV genotype 3 from pigs/deer) |
6. ALCOHOLIC-ASSOCIATED LIVER DISEASE (ALD)
Three main lesions (exist on a spectrum, can coexist):
A. Hepatic Steatosis (Alcoholic Fatty Liver)
Gross: Enlarged (up to 4-6 kg), yellow, greasy liver
Micro:
- Macrovesicular steatosis (large single fat droplet displacing nucleus to periphery) - zone 3 predominance initially
- No significant inflammation
- Reversible on abstinence (weeks)
Pathogenesis: Excess NADH from ethanol metabolism → inhibits beta-oxidation → inhibits TCA cycle → ↑ acetyl-CoA → fatty acid synthesis; also ↑ VLDL export failure; perivenular zone 3 most affected (lowest O₂ + highest CYP2E1)
B. Alcoholic Hepatitis
Gross: Enlarged, mottled liver; yellow areas of fatty change; red areas of inflammation
Micro (Hallmarks):
- Hepatocyte swelling (ballooning degeneration): Swollen hepatocytes with a clear, watery cytoplasm
- Mallory-Denk bodies (Mallory hyaline): Irregular eosinophilic intracytoplasmic inclusions composed of aggregated cytokeratins (CK8 and CK18) + ubiquitin + p62; surrounded by neutrophils (satellitosis = neutrophils surrounding Mallory-Denk body cells)
- Neutrophilic infiltrate (key difference from viral hepatitis where lymphocytes predominate)
- Hepatocyte necrosis (zone 3 predominantly)
- Megamitochondria: Giant eosinophilic mitochondria - reflect mitochondrial injury by acetaldehyde
- Perivenular fibrosis: Chicken-wire fibrosis around central vein (pericellular/sinusoidal fibrosis)
Clinical correlates:
- Fever, leukocytosis, right upper quadrant pain, jaundice
- AST:ALT ratio > 2:1 (often 2:1 to 6:1) - because: alcohol damages mitochondria → releases mitochondrial AST; alcohol depletes vitamin B₆ (PLP) required for ALT synthesis more than AST
- GGT elevated (especially sensitive for alcohol use)
- May precipitate acute-on-chronic liver failure
C. Alcoholic Cirrhosis
Gross: Initially enlarged (fatty) → later small, shrunken, nodular
- Typically micronodular (Laennec's cirrhosis) - uniform small nodules <3mm
- Deep yellow-green (bile staining), brown (hemosiderin)
Micro:
- Pericellular/sinusoidal fibrosis ("chicken-wire" pattern) around individual hepatocytes
- Perivenular fibrosis (zone 3 → porta hepatis - reverse of viral hepatitis)
- Micronodular pattern with fibrous septa
- Late stage: mixed or macronodular
- Steatosis may be absent in end-stage (fat oxidized; liver shrunken)
Clinical Stigmata of Alcoholic Liver Disease / Cirrhosis:
- Spider nevi, palmar erythema, gynecomastia, testicular atrophy (↓ liver metabolism of estrogen)
- Dupuytren's contracture, parotid enlargement
- Leukonychia (white nails)
- Jaundice, ascites, splenomegaly, varices
7. METABOLIC DYSFUNCTION-ASSOCIATED STEATOTIC LIVER DISEASE (MASLD) / NAFLD
Previously called NAFLD (Non-Alcoholic Fatty Liver Disease) - now called MASLD per 2023 nomenclature
Spectrum:
- MASL (Metabolic-Associated Steatotic Liver) = simple steatosis without inflammation or fibrosis
- MASH (Metabolic-Associated Steatohepatitis) = steatosis + inflammation + ballooning degeneration ± fibrosis (previously NASH)
- MASH cirrhosis = end-stage
- → HCC (even without cirrhosis in some cases)
Risk factors: Obesity, type 2 DM, insulin resistance, dyslipidemia, metabolic syndrome - the "multiple hit" theory
Morphology of MASH (overlaps with alcoholic hepatitis):
- Macrovesicular steatosis (panlobular but zone 3 predominant initially)
- Ballooning degeneration of hepatocytes
- Lobular inflammation (mixed - lymphocytes + macrophages + neutrophils)
- Mallory-Denk bodies (less prominent than in alcoholic hepatitis)
- Perisinusoidal fibrosis (zone 3 - "chicken-wire" = pericellular collagen) - characteristic of MASH
- NAS (NAFLD Activity Score): Steatosis (0-3) + Lobular inflammation (0-3) + Ballooning (0-2); NAS ≥5 = MASH
Key distinguishing features from alcoholic hepatitis:
- History: no significant alcohol use (<20 g/day women, <30 g/day men)
- Metabolic risk factors present
- Histologically cannot be reliably distinguished from alcoholic hepatitis (hence the clinical history is essential)
8. AUTOIMMUNE HEPATITIS (AIH)
Types (Basic Pathology):
- Type 1 (most common): ANA + ASMA (anti-smooth muscle antibody) ± anti-SLA/LP; any age
- Type 2 (mainly children/teenagers): Anti-LKM-1 (anti-liver kidney microsome 1) + anti-LC1 + anti-SLA/LP
Pathogenesis: HLA-DR3 (European), HLA-DR4 (Japanese); CD4+ T cells activate B cells → autoantibodies; CD8+ T cells at limiting plate → interface hepatitis
Morphology (Key features):
- Plasma cells in clusters (hallmark - not just single plasma cells but aggregates)
- Emperipolesis: Lymphocytes within hepatocyte cytoplasm
- Hepatocyte "rosettes": Circular arrangement of regenerating hepatocytes around dilated canaliculus
- Interface hepatitis (piecemeal necrosis) - extensive
- Confluent necrosis: Perivenular → bridging necrosis → panacinar necrosis (in severe/fulminant cases)
- Fibrosis progressive → cirrhosis at presentation in ~30% of adults
- Burnt-out cirrhosis in some (subclinical disease went unrecognized)
Treatment: Immunosuppression (prednisolone ± azathioprine) → remission in 90%; liver transplant for end-stage; 10-year survival post-transplant ~75%; recurrence in 20%
9. DRUG/TOXIN-INDUCED LIVER INJURY (DILI)
Patterns of DILI (Robbins):
| Pattern | Mechanism | Examples |
|---|---|---|
| Predictable (dose-dependent, direct toxicity) | Direct hepatotoxin; predictable at high doses | Acetaminophen (zone 3 centrilobular necrosis); carbon tetrachloride (zone 3 necrosis via free radical lipid peroxidation) |
| Unpredictable (idiosyncratic) | Drug or metabolite acts as antigen; immune-mediated; or genetically aberrant metabolism | Isoniazid, halothane, sulfonamides, alpha-methyldopa |
| Cholestatic | Impairs bile secretion/excretion | Chlorpromazine, OCPs (estrogen), anabolic steroids, cyclosporine |
| Fatty liver (steatosis) | Impairs mitochondrial fatty acid oxidation | Tetracycline (microvesicular steatosis, panlobular); valproic acid; amiodarone; methotrexate (also causes fibrosis) |
| Granulomatous | Drug-induced granulomas | Allopurinol, sulfonamides, phenytoin, beryllium |
| Vascular | Affects hepatic veins/sinusoids | Oral contraceptives (peliosis hepatis, Budd-Chiari); pyrrolizidine alkaloids (sinusoidal obstruction syndrome = hepatic veno-occlusive disease) |
| HCC | Carcinogenesis | Aflatoxin B1 |
Acetaminophen Zone 3 (Centrilobular) Necrosis:
- Zone 3 → highest CYP2E1 expression → most NAPQI generated
- Risk factors: Fasting (↓ glutathione), chronic alcohol (↑ CYP2E1 + ↓ glutathione), other CYP2E1 inducers
10. HEREDITARY HEMOCHROMATOSIS
Pathogenesis:
- Mutation in HFE gene (chromosome 6p21) - most common: C282Y homozygosity (90% of cases); H63D and compound heterozygote
- HFE protein normally downregulates intestinal iron absorption by interacting with transferrin receptor
- Mutant HFE → failure to downregulate iron absorption → net 0.5-1 g/year iron accumulation → symptoms after 20 g stored iron
- Other forms: Juvenile hemochromatosis (HJV or hepcidin mutations - presents at 20s), TfR2 mutations
Mechanisms of injury:
- Lipid peroxidation (iron-catalyzed free radical reactions)
- Stellate cell activation → collagen formation
- ROS interaction with DNA → increased risk of HCC
Morphology:
- Gross: Liver enlarged, rust-brown color
- Micro: Iron deposited in hepatocytes (periportal, zone 1 predominant initially → all zones), bile duct epithelium, Kupffer cells, pancreas (islets), heart (myocytes), joints (synovium), pituitary, skin
- Prussian blue stain (potassium ferrocyanide) = blue granules = ferric iron (hemosiderin)
- Perls' stain = synonymous
- Micronodular cirrhosis (all symptomatic patients)
- Sideronecrosis: Iron-laden hepatocytes undergoing necrosis
Clinical - Classic Triad (late-stage, now rarely seen with screening):
- Micronodular cirrhosis
- Diabetes mellitus (75-80%) = "bronze diabetes"
- Abnormal skin pigmentation (75-80%) = bronze/slate-gray skin (iron + melanin deposition)
Additional features: Cardiac arrhythmias/cardiomyopathy, hypogonadism (↓ pituitary/gonadal function), arthropathy (calcium pyrophosphate crystals, 2nd-3rd MCP joints), HCC risk (200× compared to normal)
Demographics: Males >> Females (5-7:1; menstrual blood loss compensates in premenopausal women); symptoms appear 4th-5th decade in males, later in females
Lab: ↑ Serum iron, ↑ serum ferritin, ↑ transferrin saturation (>45% = screening threshold); ↓ TIBC
11. WILSON'S DISEASE
- AR mutation in ATP7B gene (chromosome 13q14) encoding a copper-transporting ATPase in hepatocytes
- ATP7B normally transports copper:
- Into the secretory pathway for incorporation into ceruloplasmin (copper-binding protein)
- Into bile for excretion
Defect: Failure to excrete copper into bile → copper accumulates in liver → then spills into blood → deposits in brain, eye (Kayser-Fleischer rings), kidney, heart, joints
Morphology:
- Early: Fatty change (steatosis); hepatocyte glycogen nuclei (glycogenated nuclei = vacuolated nuclei); mild portal inflammation
- Progressive: Chronic hepatitis pattern → cirrhosis
- Copper staining: Rhodanine stain (orange-red copper deposits) or Orcein stain
Key Morphological feature: Hepatocyte glycogenated nuclei (nuclear glycogen accumulation - also seen in diabetes, but in Wilson's it's characteristic early finding)
Organ Involvement:
- Liver: Steatosis → chronic hepatitis → cirrhosis → acute liver failure (Coombs-negative hemolytic anemia in acute)
- Brain (Basal ganglia): Neuropsychiatric symptoms; "lenticular degeneration" - putamen cavitation; Parkinsonism, dystonia, tremor, psychiatric symptoms
- Eye: Kayser-Fleischer rings (golden-brown copper deposits in Descemet's membrane at corneal periphery) - pathognomonic in right context; seen by slit-lamp examination
- Kidney: Renal tubular acidosis (Fanconi syndrome)
- Blood: Coombs-negative hemolytic anemia during acute liver involvement
Lab: ↓ Serum ceruloplasmin (<20 mg/dL); ↑ 24-hour urine copper; ↑ liver copper (>250 µg/g dry weight on biopsy = diagnostic); serum copper may be ↓ (paradoxically - ceruloplasmin-bound fraction is low)
Treatment: D-penicillamine (chelation), trientine; zinc (reduces GI absorption by inducing intestinal metallothionein); liver transplant (curative)
12. ALPHA-1-ANTITRYPSIN (A1AT) DEFICIENCY
- AR disorder - mutation in SERPINA1 gene
- Most clinically significant mutation: PiZZ genotype (homozygous Z allele) = most common severe deficiency
- Normal A1AT: Pi*MM (PiMM); Heterozygous PiMZ: mild ↓ A1AT
- A1AT normally: serine protease inhibitor (anti-trypsin, anti-elastase) secreted by liver
Pathogenesis:
- Z-allele encodes a misfolded protein → retained in ER → forms polymers (accumulate in hepatocytes) → triggers unfolded protein response → hepatocyte injury + apoptosis
- Simultaneously: ↓ secretion of A1AT into blood → lungs unprotected from neutrophil elastase → panacinar emphysema (especially lower lobes)
Morphology:
- Periodic acid-Schiff (PAS)-positive, diastase-resistant (PAS-D) globules in hepatocyte cytoplasm (zone 1/periportal hepatocytes) = PATHOGNOMONIC finding
- The globules represent misfolded Z-type A1AT protein polymerized in ER (diastase-resistant because they are protein, not glycogen)
- Progression: steatosis → chronic hepatitis → cirrhosis → HCC
Clinical:
- Liver: Neonatal cholestasis (most common cause of neonatal cholestasis in USA), childhood liver disease, cirrhosis in adults
- Lung: Panacinar emphysema (lower lobes) in adults, especially smokers
- Risk of HCC (~35-fold in cirrhotic PiZZ patients)
- Treatment: Augmentation therapy (IV A1AT for lung disease); liver transplant (curative for liver disease and corrects A1AT deficiency because new liver has normal genotype)
13. PRIMARY BILIARY CHOLANGITIS (PBC)
- Autoimmune destruction of small intrahepatic bile ducts (interlobular bile ducts <100 µm diameter)
- Predominantly affects middle-aged women (90-95% female); F:M = 10:1
- Associated with other autoimmune conditions (Sjögren's, hypothyroidism, CREST syndrome)
Pathogenesis: Unknown trigger → CD4+ and CD8+ T cell-mediated attack against biliary epithelial cells
- Anti-mitochondrial antibodies (AMA) - present in 95% of patients; target PDC-E2 (pyruvate dehydrogenase complex E2 subunit) on mitochondrial inner membrane; highly specific; detected by immunofluorescence
Morphology (4 stages - Ludwig staging):
- Stage I (Portal stage): Portal inflammation with lymphocytes, plasma cells, eosinophils; florid duct lesion = granulomatous destruction of bile duct (lymphocytes + epithelioid granulomas surrounding bile duct) - pathognomonic
- Stage II (Periportal stage): Periportal hepatitis; ductular proliferation; progressive bile duct loss
- Stage III (Septal stage): Septal fibrosis; bridging fibrosis; progressive bile duct loss
- Stage IV (Cirrhotic stage): Biliary cirrhosis; regenerative nodules surrounded by dense fibrous tissue; copper deposits (secondary to cholestasis - impaired biliary copper excretion)
Biliary Cirrhosis Morphology:
- Micronodular cirrhosis
- Cholestasis: Bile plugs in canaliculi, bilirubin in hepatocytes
- Periductal xanthomatous change: Foamy lipid-laden macrophages (xanthoma cells) around bile ducts (from cholesterol accumulation due to impaired bile flow)
- Feathery degeneration: Periportal hepatocytes show pale, foamy, "feathery" cytoplasm due to bile salt accumulation
Clinical:
- Insidious onset of pruritus (bile acid accumulation in skin) → fatigue → jaundice
- ↑↑ ALP, ↑ GGT (cholestatic pattern)
- ↑ Serum cholesterol, IgM
- Xanthelasma, xanthomata (cholesterol deposits)
- Osteoporosis (impaired Vit D absorption + ↑ osteoclast activity from bile salt effect)
- Steatorrhea (fat malabsorption - no bile acids reaching gut)
- Treatment: UDCA (ursodeoxycholic acid) (30-40% improvement in biochemistry + slows progression) + obeticholic acid
14. PRIMARY SCLEROSING CHOLANGITIS (PSC)
- Inflammation and obliterative fibrosis of intra- and extrahepatic bile ducts of all sizes
- Strong association with IBD (Ulcerative Colitis) in 70% of cases (UC >> Crohn's)
- Male predominance (2:1); 4th-5th decade
- p-ANCA positive in ~80%; AMA negative (differentiates from PBC)
Pathogenesis: Unknown; HLA-B8, -DR2, -DR3 association; T cell-mediated bile duct injury
Morphology:
- "Onion-skin" fibrosis: Concentric periductal fibrosis around bile ducts (pathognomonic on biopsy - but biopsy has patchy distribution)
- Progressive fibrosis → bile duct strictures and dilatation → "beaded" appearance on cholangiography (MRCP/ERCP)
- "Pruned tree" appearance on ERCP = alternating strictures and dilations
- Progressive liver fibrosis → biliary cirrhosis
- Loss of bile ducts (ductopenia) in end-stage
Clinical:
- Pruritus, fatigue, jaundice; ↑ ALP, ↑ GGT
- Ascending cholangitis (biliary strictures → bacterial superinfection) - recurrent
- Cholangiocarcinoma - lifetime risk 10-20% (much higher than PBC); often arises at bifurcation (Klatskin tumor)
- Colon cancer: Increased risk due to UC association
- Treatment: Liver transplant (only cure); UDCA controversial; endoscopic dilation of dominant strictures; no effective medical therapy
15. HEPATOCELLULAR CARCINOMA (HCC)
Epidemiology
- ~5.4% of all cancers worldwide; 2nd most common cause of cancer death globally
- >85% of cases in Asia and sub-Saharan Africa (high HBV prevalence)
- Marked male predominance (8:1 in high-incidence areas)
- Peak incidence: 20-40 years (Asia) due to perinatal HBV acquisition
- In Western countries: incidence increasing due to MASLD/metabolic syndrome
Etiology and Risk Factors
| Risk Factor | Relative Risk | Notes |
|---|---|---|
| Chronic HBV | 200× | Can cause HCC even without cirrhosis |
| Chronic HCV | 100× | Almost always via cirrhosis |
| Alcoholic cirrhosis | 15-20× | Via cirrhosis |
| MASLD/MASH | 10× | Even in non-cirrhotic liver |
| Hemochromatosis | 200× | Via cirrhosis + direct iron carcinogenesis |
| Alpha-1-AT deficiency | 35× | Via cirrhosis |
| Aflatoxin B1 | Synergistic with HBV | G→T mutation in p53 codon 249; Africa/Asia |
| Wilson's disease | Mildly elevated | Via cirrhosis |
| Primary biliary cholangitis | Elevated | Via cirrhosis |
Pathogenesis
- 15-20% arise in non-cirrhotic liver (especially HBV-related)
- Common driver mutations:
- TERT promoter mutations (50-60%): ↑ telomerase → immortalization
- β-catenin (CTNNB1) activating mutations (40%): ↑ Wnt/β-catenin signaling → proliferation
- TP53 inactivating mutations (up to 60%): Loss of p53 tumor suppressor; in aflatoxin-related HCC: specific G→T transversion at codon 249
- PI3K/AKT/mTOR pathway mutations
- RAS/MAPK pathway activations
Special subtype - Fibrolamellar HCC:
- Adolescents and young adults, NO preexisting liver disease
- DNAJB1::PRKACA fusion gene → excessive protein kinase A activity
- Better prognosis than classic HCC; no elevated AFP
- Gross: Single large tumor with fibrous bands (lamellar = sheet-like)
Precursor Lesions
- Large cell change: Hepatocytes larger than normal, enlarged/pleomorphic nuclei, no ↑ N:C ratio
- Small cell change: High N:C ratio, mild nuclear hyperchromasia → more ominous precursor
- Dysplastic nodules (Low-grade and High-grade) → high-grade = immediate HCC precursor
Morphology
Gross:
- Single large mass, multiple nodules, or diffuse infiltrative growth (especially in cirrhosis)
- Typically soft, pale tan to yellow-green, vascular (highly vascular tumor - arterial)
- Bile-stained (bile production by tumor cells = only tumor that makes bile)
- Characteristic of HCC: invasion into portal vein / hepatic veins (portal vein tumor thrombus)
Micro:
- Cells resembling hepatocytes (trabecular, pseudoglandular, or acinar patterns)
- Abundant eosinophilic cytoplasm, bile canaliculi between cells
- Prominent nucleoli
- Immunohistochemistry: AFP (alpha-fetoprotein) positive; Hep Par-1 positive; Arginase-1 positive; CK8/18 positive
- Tumor cells: CK7 negative, CK20 negative (helps distinguish from metastatic adenocarcinoma)
Serum markers:
- AFP (Alpha-fetoprotein) elevated in ~70% of HCC
- Normal serum AFP: <10-15 ng/mL
- AFP >400 ng/mL in at-risk patient with liver mass = diagnostic
- AFP can also be elevated in: other liver diseases, yolk sac tumors (testicular/ovarian)
- PIVKA-II (Des-γ-carboxyprothrombin) = sensitive and specific marker; used in Asia
Surveillance
- 6-monthly ultrasound ± AFP in cirrhotic patients and HBV carriers with high risk factors
- Contrast CT/MRI: arterial phase enhancement + portal venous washout = LI-RADS 5 = HCC (no biopsy needed for classic pattern)
16. CHOLANGIOCARCINOMA (CCA)
- Malignant neoplasm derived from cholangiocytes (bile duct epithelium)
| Type | Location | % | Risk factors |
|---|---|---|---|
| Intrahepatic CCA | Within liver parenchyma | 10-15% | PSC, HCV, HBV, MASLD, liver flukes (Clonorchis sinensis, Opisthorchis viverrini), Caroli disease |
| Perihilar CCA (Klatskin tumor) | Confluence of R and L hepatic ducts | ~50% | PSC (major risk), HBV, HCV |
| Distal CCA | Extrahepatic bile duct distal to cystic duct | ~35% | Choledochal cysts, CBD stones |
Morphology:
- Dense desmoplastic reaction (abundant fibrous stroma → hard scirrhous tumor)
- Gland-forming mucin-secreting adenocarcinoma
- IHC: CK7 positive, CK19 positive, CK20 negative (intrahepatic CCA); AFP negative; CA 19-9 elevated (serum)
Clinical:
- Jaundice (obstructive - perihilar, distal); or incidental liver mass (intrahepatic)
- Poor prognosis (most unresectable at diagnosis)
- Perihilar CCA: biliary stenting; liver transplant in select PSC patients with perihilar CCA
17. VASCULAR DISORDERS OF THE LIVER
Budd-Chiari Syndrome (Hepatic Vein Outflow Obstruction)
- Thrombosis or occlusion of hepatic veins (±IVC)
- Causes: Polycythemia vera (most common), hypercoagulable states, oral contraceptives, pregnancy, myeloproliferative disorders, paroxysmal nocturnal hemoglobinuria, Behçet's disease, membranous webs of IVC (in Asia/Africa)
- Morphology:
- Gross: Liver enlarged, congested, purple-red
- Micro: Severe centrilobular congestion (zone 3) → zone 3 hepatocyte necrosis → fibrosis → "congestive cirrhosis"
- Caudate lobe hypertrophy (pathognomonic): Caudate lobe drains directly into IVC independently → spared from venous obstruction → compensatory hypertrophy (visible on imaging)
- Clinical: Acute: abdominal pain, hepatomegaly, ascites; Chronic: progressive cirrhosis; some present with fulminant hepatic failure
Cardiac Hepatopathy (Congestive Heart Failure)
- Chronically elevated central venous pressure → hepatic venous congestion
- Morphology: "Nutmeg liver" (gross) = red centrilobular congestion (zone 3) contrasting yellow periportal parenchyma (fattyl change)
- Micro: Zone 3 hepatocyte necrosis → "cardiac cirrhosis" (rare, but possible in long-standing CHF)
- Centrilobular fibrosis pattern (unlike other forms of cirrhosis which start in portal tracts)
Sinusoidal Obstruction Syndrome (SOS) / Hepatic Veno-Occlusive Disease (HVOD)
- Obstruction at sinusoidal and centrilobular venule level (NOT hepatic vein thrombosis)
- Causes: High-dose chemotherapy (busulfan, cyclophosphamide) before bone marrow transplant; pyrrolizidine alkaloids (herbal teas, Jamaican "bush tea"); radiation
- Endothelial cell damage → edema → fibrin deposition → sinusoidal and centrilobular venule narrowing
- Morphology: Zone 3 congestion and necrosis without hepatic vein thrombosis; centrilobular fibrosis
Portal Vein Thrombosis (PVT)
- Extrahepatic PVT causes: Cirrhosis (50%), intra-abdominal inflammation (appendicitis, peritonitis), malignancy (HCC invades portal vein), coagulation disorders
- Results in: Portal hypertension with splenomegaly; can form cavernous transformation (collateral vessels around thrombosed portal vein = cavernoma)
- Acute PVT: may cause bowel ischemia if SMV involved
18. BENIGN LIVER TUMORS
| Tumor | Features | Key Points |
|---|---|---|
| Hepatic Hemangioma | Most common benign liver tumor; cavernous vascular spaces lined by flat endothelium; usually incidental on imaging; well-defined, hyperechoic on US; "fill-in" enhancement on MRI | No malignant potential; biopsy risk of bleeding → avoid |
| Hepatocellular Adenoma | Benign hepatocyte proliferation; well-differentiated hepatocytes with absent portal tracts and bile ducts; risk factors: OCP use (estrogen-dependent), anabolic steroids, glycogen storage disease type Ia; rare but significant because may bleed or undergo malignant transformation (especially β-catenin mutated subtype) | HNF1α subtype = steatotic; β-catenin subtype = high malignant risk; inflammatory subtype = serum CRP elevated |
| Focal Nodular Hyperplasia (FNH) | Non-neoplastic hyperplastic nodule; central fibrous scar with radiating septa; prominent thick-walled arteries in the scar (arteriovenous malformation-like); no portal tracts within nodules but bile ductules in fibrous septa; Kupffer cells present (positive on sulfur colloid nuclear scan) | Not a true neoplasm; "stealth lesion" → no malignant potential; characteristic "spoke-wheel" vascularity on Doppler; no treatment needed |
19. SUMMARY TABLE - KEY HISTOLOGICAL FINDINGS IN LIVER PATHOLOGY
| Disease | Zone affected | Key Histological Finding | Special Stain |
|---|---|---|---|
| Acetaminophen toxicity | Zone 3 | Centrilobular necrosis | - |
| Yellow fever / eclampsia | Zone 2 (midzone) / Zone 1 | Midzone necrosis / periportal necrosis | - |
| Phosphorus / eclampsia / preeclampsia | Zone 1 | Periportal necrosis | - |
| Cardiac failure / Budd-Chiari | Zone 3 | Centrilobular congestion and necrosis | - |
| Alcoholic hepatitis | Zone 3 | Mallory-Denk bodies, neutrophil satellitosis, steatosis | - |
| Alcoholic cirrhosis | Zone 3 → all | Micronodular, chicken-wire fibrosis | Masson's trichrome |
| Viral hepatitis (acute) | Lobular (diffuse) | Acidophil bodies (Councilman bodies), lobular disarray | - |
| HBV (chronic) | Zone 1 | Ground-glass hepatocytes, sanded nuclei | Orcein, Victoria blue |
| HCV (chronic) | Portal | Lymphoid follicles, bile duct damage, steatosis | - |
| Autoimmune hepatitis | Portal/interface | Plasma cell clusters, rosettes, emperipolesis | - |
| PBC | Portal | Florid duct lesion, granulomatous duct destruction, copper | Rhodanine, Orcein |
| PSC | Portal | Onion-skin periductal fibrosis, bile duct strictures | Trichrome |
| Hemochromatosis | Zone 1 → all | Hemosiderin in hepatocytes, Kupffer cells, bile ducts | Prussian blue / Perls' |
| Wilson's disease | Any | Glycogenated nuclei, steatosis, copper deposits | Rhodanine |
| Alpha-1-AT deficiency | Zone 1 (periportal) | PAS-positive diastase-resistant globules in hepatocytes | PAS + diastase |
| MASH (NAFLD/NASH) | Zone 3 | Macrovesicular steatosis, ballooning, Mallory-Denk, perisinusoidal fibrosis | Trichrome |
| HCC | N/A | Trabecular pattern, bile production, AFP+ | AFP, Hep Par-1, Arginase-1 |
| Cholangiocarcinoma | N/A | Gland-forming adenocarcinoma, dense desmoplasia | CK7+, CK19+, CK20- |
Sources: Robbins & Cotran Pathologic Basis of Disease (9780443264528, pages 769-869), Robbins & Kumar Basic Pathology (9780323790185, pages 533-620)
That covers the complete pathology of the liver - from hepatic failure, cirrhosis pathogenesis, portal hypertension, all hepatotropic viruses (HAV-HEV with morphology, serology, chronicity), alcoholic liver disease, MASLD/NAFLD, autoimmune hepatitis, DILI, hereditary hemochromatosis, Wilson's disease, alpha-1-antitrypsin deficiency, PBC, PSC, HCC, cholangiocarcinoma, vascular disorders, benign tumors, and a complete special stain quick-reference table.
Next topics:
- Microbiology (Hepatitis viruses in detail - Jawetz + Murray)
- Pharmacology (Antivirals, drugs for complications - Goodman & Gilman + Katzung)
- Medicine (Clinical management - Harrison's 22nd Ed)
- Surgery (Hepatic resection, portal hypertension surgery - Schwartz/Sabiston)
Micro
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Here is the complete textbook-level microbiology of the liver, synthesized from Murray Medical Microbiology 9e, Jawetz Medical Microbiology 28e, and Sherris & Ryan Medical Microbiology 8e.
MICROBIOLOGY OF THE LIVER - Full Textbook-Level Notes
OVERVIEW: THE FIVE HEPATOTROPIC VIRUSES
| Feature | HAV | HBV | HCV | HDV | HEV |
|---|---|---|---|---|---|
| Common name | "Infectious hepatitis" | "Serum hepatitis" | "Non-A, non-B posttransfusion" | "Delta agent" | "Enteric non-A, non-B" |
| Family | Picornaviridae | Hepadnaviridae | Flaviviridae | Viroid-like | Hepeviridae |
| Genome | (+) ssRNA, 7.5 kb | Partially dsDNA, 3.2 kb | (+) ssRNA, 9.4 kb | Circular (-) ssRNA | (+) ssRNA |
| Envelope | No | Yes | Yes | Yes (uses HBsAg) | No |
| Transmission | Fecal-oral | Parenteral, sexual, perinatal | Parenteral, (sexual low) | Parenteral | Fecal-oral |
| Incubation | 15-50 days | 45-160 days | 14-180+ days | 15-64 days | 15-50 days |
| Onset | Abrupt | Insidious | Insidious | Abrupt | Abrupt |
| Chronicity | Never | 5-10% (adults); 90% (neonates) | 70-90% | Co-inf: 5-10%; Superinf: 90-100% | Never (except immunocompromised) |
| Carrier state | No | Yes | Yes | Yes | No |
| HCC association | None | Yes (major) | Yes | Yes (via cirrhosis) | None |
| Mortality | <0.5% | 1-2% | ~4% | High to very high | 1-2%; 20% in pregnancy |
| Vaccine | Yes (inactivated) | Yes (recombinant) | None | HBV vaccine prevents HDV | None (in many countries) |
1. HEPATITIS A VIRUS (HAV)
Taxonomy and Structure
- Family: Picornaviridae, genus: Hepatovirus (only hepatotropic picornavirus)
- Particle: 27-32 nm, spherical, icosahedral capsid (non-enveloped)
- Genome: Positive-sense ssRNA, 7.5 kb, linear; single open reading frame
- One serotype (only one; cross-protective immunity worldwide)
- Seven genotypes (I-VII) based on variable sequence at 1D/2A junction; genotype I is most prevalent in humans
- Proteins: VP1, VP2, VP3, VP4 = capsid; VPg = genome-linked protein; 2Apro/3Cpro = proteases; 3Dpol = RNA-dependent RNA polymerase
Resistance to Physical/Chemical Agents (Jawetz)
HAV is unusually stable compared to other picornaviruses:
- Stable at: pH 1.0 for 2 hours; 20% ether; 60°C for 1 hour; -20°C storage for years
- Destroyed by: Autoclaving (121°C, 20 min); boiling for 5 min; dry heat 180°C; UV irradiation (1 min at 1.1 W); formalin (1:4000, 3 days at 37°C); chlorine (10-15 ppm, 30 min); sodium hypochlorite 1:100
- Heating food to >85°C for 1 minute inactivates HAV
- Practical implication: Normal household disinfectants may be insufficient; bleach required for contaminated surfaces
Receptor and Cell Entry
- HAVcr-1 (TIM-1/KIM-1) on hepatocyte surface is the primary receptor for cell entry
- HAV has a very limited cell tropism (essentially only hepatocytes)
- In vitro: Various primate cell lines support replication, but no cytopathic effects; slow adaptation required for fresh isolates
Replication and Pathogenesis
- HAV replicates in the cytoplasm of hepatocytes
- Shed in bile → high concentrations in feces (peak infectivity in last 2 weeks of incubation period and first week of symptoms)
- NOT cytopathic directly - liver injury is immunologically mediated:
- Cytotoxic T lymphocytes (CTL/CD8+) recognize HAV antigens on hepatocyte surface → hepatocyte lysis
- NK cells contribute to early non-specific killing
- Interferon (IFN-α/β) production by infected cells limits viral spread
- After symptoms appear, viral shedding falls rapidly (humoral immunity clears virus)
Clinical Course
- Incubation: 15-50 days (mean 28 days)
- Prodromal phase (pre-icteric, 1-2 weeks): Fever, malaise, anorexia, nausea, RUQ discomfort; dark urine; liver tender on palpation
- Icteric phase: Jaundice appears (if symptomatic); liver enlargement; most adults are icteric; children often anicteric/subclinical
- Convalescence: 1-3 months; complete recovery
- No chronicity (no persistent infection)
- Fulminant hepatitis (massive hepatic necrosis): 0.1-0.3%; higher risk in patients with pre-existing liver disease (chronic HBV, HCV, cirrhosis)
- Relapsing hepatitis: 10-15% have a second wave of disease after apparent recovery - self-limited
- Cholestatic hepatitis: Prolonged cholestasis with pruritus; responds to corticosteroids
Serology and Diagnosis (Murray)
- Anti-HAV IgM: Appears at onset of symptoms; persists 3-6 months → diagnosis of acute infection
- Anti-HAV IgG: Appears weeks after IgM; persists lifelong → indicates past infection or vaccination → marker of immunity
- Stool HAV antigen: Detectable 1-2 weeks before and up to 1-2 weeks after onset of symptoms
- HAV RNA by RT-PCR: Most sensitive; used in outbreak investigation and blood safety screening
Prevention
- Inactivated HAV vaccine (Havrix, Vaqta): 2 doses, 6-12 months apart; nearly 100% efficacy; long-lasting immunity
- Immunoglobulin (IG): Post-exposure prophylaxis within 2 weeks of exposure; also used for travelers to endemic areas if too close to departure for vaccine
- Vaccine + IG: For highest-risk exposed individuals
- Sanitation: Proper sewage disposal, safe water supply, hand hygiene; HAV survives on environmental surfaces
2. HEPATITIS B VIRUS (HBV)
Taxonomy and Structure (Murray + Jawetz)
- Family: Hepadnaviridae (Hepatitis DNA viruses); Genus: Orthohepadnavirus
- Animal relatives: Woodchuck hepatitis virus (WHV), Ground squirrel hepatitis virus (GSHV), Duck hepatitis B virus (DHBV) - all have limited host ranges; useful animal models
- Dane particle: Complete 42 nm virion; most complex hepatitis virus
- Outer lipid envelope containing three forms of HBsAg (S, M, L)
- Inner 27 nm icosahedral nucleocapsid (HBcAg)
- Inside core: partially double-stranded circular DNA genome, 3200 bp (3.2 kb); covalently closed circular DNA (cccDNA) in nucleus; reverse transcriptase (RT/RNase H = P protein)
- HBsAg particles in serum: Two additional non-infectious forms produced in massive excess:
- 22 nm spheres (most abundant - 10,000× excess over Dane particles): composed of HBsAg S protein; not infectious; immunogenic → used for original plasma-derived vaccine
- Filamentous/tubular forms (22 nm diameter, >200 nm long): S, M, L HBsAg
- These excess HBsAg particles serve as decoys for the immune system
HBsAg Proteins (Murray)
Three forms encoded by the S gene (read in same frame, different start codons):
- S protein (gp27/24-27 kDa): Small; main component of 22-nm particles; completely contained within M and L
- M protein (gp36/33-36 kDa): Medium; contains preS2 + S
- L protein (gp42/39-42 kDa): Large; contains preS1 + preS2 + S; essential for viral attachment to hepatocytes (preS1 binds NTCP = sodium-taurocholate co-transporting polypeptide on hepatocyte membrane)
Genome Organization - 4 Open Reading Frames (ORFs)
All ORFs overlap (highly compact genome - each nucleotide is part of at least one ORF):
| ORF | Encodes | Function |
|---|---|---|
| S gene | HBsAg (L, M, S proteins) | Envelope; viral attachment (preS1-NTCP); vaccine antigen |
| C gene | HBcAg + HBeAg | Core capsid (HBcAg); secreted marker of replication (HBeAg via precore sequence) |
| P gene (largest) | P protein (reverse transcriptase/RNase H/DNA polymerase) | Viral replication; priming negative-strand DNA synthesis; removal of RNA template |
| X gene | HBx protein (transcriptional transactivator) | Activates viral and cellular gene promoters; implicated in HCC carcinogenesis (activates oncogenes, inhibits p53) |
HBV Replication Cycle (Jawetz - crucial for antiviral targets)
- Attachment: L-HBsAg (preS1 domain) binds NTCP (SLC10A4) on hepatocyte - the functional receptor identified in 2012
- Fusion: Viral envelope fuses with hepatocyte membrane → nucleocapsid enters cytoplasm
- Nuclear transport: Core particle transported to nucleus → releases genome
- cccDNA formation: Partially double-stranded DNA → host enzymes repair to form covalently closed circular DNA (cccDNA) - the viral "minichromosome" - template for all viral RNAs; persists lifelong in hepatocyte nucleus; cannot be eradicated by current antivirals
- Transcription: cccDNA → four viral mRNAs (by host RNA polymerase II):
- 3.5 kb pregenomic RNA (pgRNA) - most important: packaged in new cores; serves as template for reverse transcription
- 2.4 kb, 2.1 kb (for HBsAg forms), 0.7 kb (for HBx)
- Packaging: pgRNA + P protein + cellular proteins → packaged into new cores (by HBcAg)
- Reverse transcription (by P protein) inside core:
- P protein primes its own reverse transcription using a 5' hairpin of pgRNA as template → negative-strand DNA
- RNase H activity of P degrades the pgRNA template
- Positive-strand DNA synthesis (incomplete → partially ds DNA = mature genome)
- Envelopment: Mature core buds through ER/Golgi acquiring HBsAg envelope → secreted as Dane particle
- OR core re-enters nucleus → amplifies cccDNA pool
- cccDNA persistence: 5-50 copies per cell; responsible for viral persistence and rebound after stopping antivirals
HBV Stability
- Unusually stable for an enveloped virus - resists ether, low pH, freezing, moderate heating
- Infectious on surfaces at room temperature for ≥7 days
- HBV is NOT readily inactivated by detergents (unlike most enveloped viruses)
- Disinfect with 10% bleach; autoclave for instruments
HBV Antigens, Antibodies, and Serology - Complete
| Marker | Nature | Meaning |
|---|---|---|
| HBsAg | Envelope glycoprotein secreted to blood | First to appear in acute infection (before symptoms); persistence >6 months = chronic HBV; carrier state |
| Anti-HBs | Antibody to HBsAg | After recovery or vaccination; marker of immunity; last to appear in recovery |
| HBcAg | Core (nucleocapsid) protein | NOT detectable in serum (stays inside virus/hepatocytes); detected on liver biopsy (IHC) |
| Anti-HBc IgM | IgM antibody to HBcAg | Appears 1-2 weeks after HBsAg; high titer = acute infection; sole marker in window period |
| Anti-HBc IgG | IgG antibody to HBcAg | Persists lifelong; indicates past or current infection; alone (with negative HBsAg and positive anti-HBs) = recovery |
| HBeAg | Secreted precore protein | Active viral replication + high infectivity (correlates with HBV DNA) |
| Anti-HBe | Antibody to HBeAg | Seroconversion = ↓ replication; lower infectivity; but precore mutant virus is HBeAg-negative despite active replication |
| HBV DNA | Viral genome in blood | Best direct measure of replication; quantitative PCR for monitoring treatment |
Serological profiles:
| Phase | HBsAg | Anti-HBs | Anti-HBc | HBeAg | Anti-HBe | HBV DNA |
|---|---|---|---|---|---|---|
| Incubation | + | - | - | + | - | +++ |
| Acute symptomatic | + | - | IgM + | + | - | ++ |
| Window period | - | - | IgM + | - | +/- | +/- |
| Recovery | - | + | IgG + | - | + | - |
| Chronic (replicating/HBeAg+) | + | - | IgG + | + | - | +++ |
| Chronic (low replication/HBeAg-) | + | - | IgG + | - | + | +/- |
| Vaccination | - | + | - | - | - | - |
| "Isolated anti-HBc" | - | - | IgG + | - | - | - |
"Isolated anti-HBc": HBsAg negative, anti-HBs negative, anti-HBc IgG positive. Seen in: (1) window period, (2) remote past infection (anti-HBs fell below detection), (3) occult HBV (low-level HBV DNA in liver without detectable HBsAg)
HBV Genotypes
- 10 genotypes (A-J) based on >8% nucleotide divergence
- Clinically relevant:
- Genotype A: Europe, North America; responds better to pegylated interferon
- Genotype B, C: Asia; Genotype C associated with higher HCC risk
- Genotype D: Mediterranean, India; more resistance mutations
Precore Mutant HBV
- Mutation in precore region (stop codon at codon 28: G1896A) → cannot produce HBeAg
- HBeAg-negative, anti-HBe positive, but HBV DNA positive (ongoing active replication)
- More aggressive disease; common in Mediterranean (genotype D), Asia (genotype B/C)
- More relapses on antiviral therapy
Pathogenesis of Liver Injury
- HBV is NOT directly cytopathic at low-to-moderate levels of replication
- Liver injury is primarily immunologically mediated:
- CD8+ CTLs recognize HLA class I + viral peptides on hepatocyte surface → hepatocyte lysis
- CD4+ T helper cells (recognizing HBcAg/HBeAg peptides) amplify the immune response
- In immunocompetent adults: vigorous immune response → symptomatic hepatitis BUT effective viral clearance (95%)
- In neonates: immature immune response → no effective clearance → chronic infection (90%) with minimal acute liver injury
- In chronic infection: persistent lower-level immune response → continuous hepatocyte turnover → cirrhosis → HCC
- HBx protein plays a direct carcinogenic role: activates oncogenes (c-myc, c-fos), inhibits TP53, activates Ras/MAPK and Wnt/β-catenin pathways
- HBV DNA integrates into host genome in chronic infection → insertional mutagenesis → additional HCC risk
Prevention and Vaccines
- Recombinant HBsAg vaccine (Engerix-B, Recombivax HB): S gene expressed in Saccharomyces cerevisiae → HBsAg self-assembles into 20-nm VLPs → alum adjuvant
- 3-dose series (0, 1, 6 months): >95% seroconversion
- Universal infant vaccination (neonatal dose within 12-24h if mother HBsAg+) - critical for interrupting perinatal transmission
- Heplisav-B (third-generation): Yeast-derived HBsAg + CpG-ODN (TLR-9 agonist) adjuvant → only 2 doses, 1 month apart; better immunogenicity
- HBIG (Hepatitis B Immune Globulin): Passive immunization; for post-exposure prophylaxis (needlestick, perinatal exposure); given within 24h; combined with vaccine for neonates of HBsAg+ mothers
3. HEPATITIS C VIRUS (HCV)
Taxonomy and Structure (Murray + Jawetz)
- Family: Flaviviridae, genus: Hepacivirus (only member of this genus)
- Particle: 30-60 nm, enveloped, spherical
- Genome: Positive-sense ssRNA, 9.4 kb; single long open reading frame (polyprotein precursor)
- The genome encodes 10 proteins (3 structural + 7 non-structural):
- Structural: Core (nucleocapsid protein, C), E1 glycoprotein, E2 glycoprotein
- Non-structural: p7 (ion channel/viroporin), NS2, NS3 (serine protease + helicase/NTPase), NS4A (NS3 cofactor), NS4B, NS5A (membranous web formation, replication complex), NS5B (RNA-dependent RNA polymerase - RdRp)
HCV Genome Organization
5'UTR - C - E1 - E2 - p7 - NS2 - NS3 - NS4A - NS4B - NS5A - NS5B - 3'UTR
Structural | Non-structural
- 5' UTR: Contains IRES (internal ribosome entry site) - cap-independent translation
- E1, E2: Highly variable envelope glycoproteins; hypervariable region 1 (HVR-1) of E2 = main target of neutralizing antibodies → rapid mutation → immune escape → explains why vaccine development is so difficult
- NS3 protease: Target of first-generation DAAs (boceprevir, telaprevir; now superseded by NS3/4A inhibitors like simeprevir, grazoprevir, voxilaprevir)
- NS5A: Target of NS5A inhibitors (daclatasvir, ledipasvir, velpatasvir, pibrentasvir)
- NS5B (RdRp): Target of nucleotide analog NS5B inhibitors (sofosbuvir)
HCV Entry Receptors
HCV binds multiple surface receptors on hepatocytes and B lymphocytes:
- CD81 (tetraspanin): Primary receptor for E2
- SR-B1 (Scavenger Receptor class B type I): Binds E2
- Claudin-1, Occludin: Tight junction proteins used as co-receptors (post-attachment entry)
- LDLR (LDL receptor): HCV coats itself with VLDL/LDL (lipoviroparticle) → entry via lipoprotein receptor
- This multi-receptor strategy makes receptor-targeted therapy very difficult
HCV Genotypes
- 7 major genotypes (clades/1-7) and >100 subtypes
- Clades differ from each other by 25-35% at nucleotide level; subtypes by 15-25%
- Within each infected patient: quasispecies = swarm of closely related but slightly different sequences generated by NS5B error-prone replication → rapid selection for drug resistance mutations; helps explain immune evasion
| Genotype | Geographic Distribution | Clinical Note |
|---|---|---|
| 1a, 1b | North America, Europe, Japan | Most common globally; 1b more common in Europe/Japan; slightly harder to treat with older IFN-based regimens; Pan-genotypic DAAs equally effective |
| 2 | Japan, Europe, South America | Good IFN response historically |
| 3 | India, Pakistan, Southeast Asia | Associated with steatosis (direct viral effect); higher HCC risk; slightly harder to treat |
| 4 | Middle East, Africa | Common in Egypt (unsafe medical injections - highest prevalence globally) |
| 5 | South Africa | |
| 6 | Southeast Asia |
HCV Replication
- Entry (as above) → cytoplasmic RNA translation via IRES → polyprotein (3011 AA) → processed by NS2-NS3 viral protease + host signal peptidase → all 10 proteins
- Replication complex forms on membranous web (endoplasmic reticulum-derived) = "membranous web" = assembled by NS4B
- NS5B replicates RNA genome through negative-strand intermediate (no DNA; no cccDNA → different from HBV; can be cured)
- Assembly at ER → budding into ER lumen → transit through Golgi → secreted as enveloped virion
- No nuclear phase; no cccDNA → explains why cure (SVR) is achievable with DAAs
Clinical Course of HCV
- Most new infections (70-80%) are subclinical (no symptoms or mild non-specific illness)
- Acute symptomatic phase: Mild, non-specific (malaise, nausea); jaundice in <25%
- Chronicity: 70-90% fail to clear → chronic HCV
- 20-30% of chronic HCV → cirrhosis over 20-30 years
- Of those with cirrhosis: ~2-4% per year risk of HCC
- Extrahepatic manifestations (immunological, from immune complex deposition):
- Mixed cryoglobulinemia (Type II - monoclonal RF + polyclonal IgG) → vasculitis, glomerulonephritis, purpura, arthralgias
- Membranoproliferative glomerulonephritis (MPGN)
- B-cell non-Hodgkin's lymphoma (HCV drives B cell proliferation via CD81)
- Porphyria cutanea tarda (PCT)
- Autoimmune thyroiditis; Sjögren's-like syndrome; lichen planus
Diagnosis
- Anti-HCV ELISA (3rd generation): Screening test; detects antibodies to multiple HCV antigens (core, NS3, NS4, NS5); does NOT distinguish acute, chronic, or cleared infection; false negatives in acute infection (window period up to 8-12 weeks after exposure)
- HCV RNA by RT-PCR (quantitative/qualitative): Confirms active infection; measures viral load; monitors treatment response
- Qualitative: limit of detection ~10-15 IU/mL
- Quantitative: expressed as IU/mL; correlates with treatment response
- HCV core antigen assay: Detectable earlier than anti-HCV; correlates with viral load; useful where PCR unavailable
- HCV genotyping: Guides choice of DAA regimen and treatment duration
- Liver fibrosis assessment: FIB-4 score, FibroScan (transient elastography), or liver biopsy (Metavir staging F0-F4)
No Vaccine
- High mutation rate of E1/E2 (hypervariable region) → no cross-protective immune response to different quasispecies
- Over 100 subtypes; immune response to one does not protect against another
- Multiple DAAs now achieve >95% SVR → focus on treatment as prevention
4. HEPATITIS D VIRUS (HDV)
Structure and Unique Biology (Murray + Jawetz)
- Defective RNA virus - an "obligate parasite" of HBV
- Particle: 35-37 nm; has its own antigen (HDAg = delta antigen) but borrows HBsAg as its outer envelope
- Genome: Circular, negative-sense ssRNA ~1700 nucleotides (smallest animal virus genome); resembles plant viroid or satellite RNA
- HDV RNA is self-complementary (rod-like secondary structure) → resembles plant viroids
- HDAg: Two forms - small HDAg (p24) promotes replication; large HDAg (p27) inhibits replication and is required for assembly (addition of prenylation signal)
- Replication: Uses host RNA polymerases (hijacks cellular Pol II and Pol I for replication) → no viral RNA polymerase encoded
HDV and HBV Dependence
- HDV absolutely requires HBsAg for:
- Its own envelope (wraps its nucleoprotein in HBsAg coat)
- Cell entry (preS1 domain of HBsAg binds NTCP)
- Therefore:
- HDV only infects individuals already infected with HBV
- HBV vaccination prevents HDV (if no HBV, no HDV)
- HBV antivirals that suppress HBsAg may reduce HDV replication
Forms of HDV Infection
- Co-infection (HDV + HBV simultaneously, both acute):
- Presents as severe acute hepatitis with biphasic transaminase elevation (two peaks)
- Usually self-limited (only 5-10% chronic); immune system clears both viruses together
- Risk of fulminant hepatic failure higher than HBV alone
- Superinfection (acute HDV infecting a chronic HBV carrier):
- 90-100% chronic HDV
- Accelerated progression to cirrhosis; higher risk of fulminant hepatitis
- 5-10 years earlier development of cirrhosis compared to HBV alone
- Worst prognosis of any hepatitis combination
Diagnosis
- Anti-HDV IgM and IgG: ELISA; IgM indicates active HDV
- HDV RNA by PCR: Quantitative; most sensitive; best for monitoring
- HDAg in liver: Immunostaining; nuclear predominance
5. HEPATITIS E VIRUS (HEV)
Taxonomy and Structure
- Family: Hepeviridae, genus: Orthohepevirus; previously incorrectly classified with Caliciviruses
- Particle: 27-34 nm, spherical, non-enveloped (naked capsid, like HAV)
- Genome: Positive-sense ssRNA, 7.2 kb; 3 open reading frames:
- ORF1: Non-structural proteins (methyltransferase, helicase, RdRp)
- ORF2: Capsid protein (viral antigen; target of antibody)
- ORF3: Small multifunctional protein (involved in virion release)
HEV Genotypes and Epidemiology
| Genotype | Hosts | Distribution | Transmission | Notes |
|---|---|---|---|---|
| HEV-1 | Humans only | Asia, Africa, Central America | Contaminated water (epidemic) | Most pathogenic in pregnancy |
| HEV-2 | Humans only | Mexico, Africa | Contaminated water | |
| HEV-3 | Humans + pigs, deer | Europe, USA (zoonotic) | Undercooked pork/game meat; sporadic | Causes chronic HEV in immunocompromised |
| HEV-4 | Humans + pigs | China | Similar to HEV-3 |
- HEV-1 and HEV-2: Epidemic; waterborne; developing countries; major outbreaks after flooding (India, Bangladesh, Pakistan, Myanmar, Nepal, Sudan)
- HEV-3 and HEV-4: Zoonotic; sporadic; developed countries; pigs are the main reservoir
Clinical Features
- Incubation: 15-50 days
- Acute, self-limited hepatitis; clinically indistinguishable from HAV in most patients
- Special risk: pregnant women (especially 3rd trimester):
- HEV-1 infection → fulminant hepatitis in 20-25% of pregnant women
- Mechanism: hormonal immune modulation in pregnancy (elevated progesterone suppresses Th1 responses) + placental HEV replication → vertical transmission → fetal infection + spontaneous abortion
- This is unique to HEV-1 (HEV-3 is much less virulent in pregnancy)
- Chronic HEV: Only in immunocompromised hosts (solid organ transplant recipients, HIV patients with CD4 < 200, hematologic malignancies on chemotherapy):
- HEV-3 and HEV-4 responsible
- May lead to rapid progression to cirrhosis (within 2-3 years) if untreated
- Treatment: Reduce immunosuppression; ribavirin (12-24 weeks; cure rate ~80%)
- Extrahepatic: HEV associated with neurological manifestations (Guillain-Barré syndrome, neuralgic amyotrophy/brachial neuritis, meningitis)
- HEV-3 in pig slaughterhouse workers → occupational exposure
Diagnosis
- Serum anti-HEV IgM (acute)
- Serum anti-HEV IgG (past infection/immunity)
- HEV RNA by RT-PCR (blood or stool): most sensitive in early acute phase; essential for diagnosis in immunocompromised (serology may be negative due to impaired antibody response)
HEV Vaccine
- Hecolin (HEV 239): Recombinant capsid protein vaccine; licensed and used in China (2012); highly efficacious
- Not yet universally available in most countries (as of 2026)
6. HEPATITIS G VIRUS (HGV/GBV-C)
(Murray Medical Microbiology - Hepatitis G Virus heading at line 2229)
- Flavivirus related to HCV; now called GBV-C (GB virus C)
- Transmitted parenterally; common in blood donors
- Despite early concern, HGV/GBV-C does NOT cause hepatitis
- May actually have a protective effect against HIV progression (immune modulation)
- No treatment required; largely of historical/research interest
PART B: NON-VIRAL MICROBIOLOGY OF THE LIVER
7. HEPATIC PARASITES - TREMATODES (FLUKES)
A. Clonorchis sinensis (Chinese Liver Fluke) - Sherris 8e
Classification:
- Phylum Platyhelminthes, Class Trematoda
- Related flukes: Opisthorchis viverrini (Southeast Asia - Thailand, Laos), Opisthorchis felineus (Eastern Europe), Fasciola hepatica (sheep liver fluke - worldwide)
Morphology:
- Small, flat, leaf-shaped adult worm: 5 × 15 mm
- Cone-shaped anterior pole; large oral sucker + ventral sucker
- Two deeply lobular testes in posterior third (distinguishing feature)
- Hermaphroditic
- Eggs: 15 × 30 µm (tiny = smallest helminth egg in clinical practice); urn-shaped with opercular rim (shoulder at junction) + posterior knob; yellow-brown, refractile
Life Cycle (Heteroxenous - 3 hosts):
- Definitive host (humans, cats, dogs, rats, pigs): Adult worms live in intrahepatic bile ducts for up to 50 years; produce ~2000 eggs/day → eggs exit in bile → pass in feces
- First intermediate host (freshwater snail): Eggs ingested → miracidium → sporocyst → redia → cercariae → released into water
- Second intermediate host (freshwater fish): Cercariae penetrate fish tissues → encyst as metacercariae (infective stage for humans)
- Human infection: Eating raw, undercooked, fermented, dried, salted, or pickled freshwater fish → metacercariae released in duodenum → ascend common bile duct → reach second-order intrahepatic bile ducts → mature to adults over 30 days
Epidemiology:
- Endemic in: Korea, Japan, Taiwan, Red River Valley of Vietnam, Guangdong province (China), Hong Kong
- Up to 35 million infected worldwide
- Entire adult population may be infected in some Chinese villages
- Perpetuated by fertilizing fish ponds with human feces (improving sanitation → decreasing prevalence, but slowly due to long lifespan)
Pathogenesis and Disease:
- Adult worm in bile duct → feeds on biliary mucosal secretions → induces epithelial hyperplasia, adenoma formation, periductal inflammation
- Light infections: usually asymptomatic
- Heavy infections (worm loads 500-1000 from repeated reinfection):
- Bile stone formation (dead worm fragments + bile pigments)
- Biliary obstruction → secondary bacterial cholangitis → bacteremia/endotoxemia
- Asymptomatic biliary carriage of Salmonella Typhi (worms harbor bacteria in bile ducts)
- Occasional extension into pancreatic ducts → pancreatitis
- Cholangiocarcinoma (most important long-term complication) - WHO Group 1 carcinogen
- Mechanism: chronic biliary epithelial irritation → metaplasia → dysplasia → malignancy
- Opisthorchis viverrini carries the highest cholangiocarcinoma risk of all liver flukes
Diagnosis:
- Stool microscopy: identify characteristic eggs (shoulder + posterior knob)
- Duodenal aspirate: more sensitive
- Serology: ELISA (less specific due to cross-reactivity)
- Imaging (US/CT): dilated intrahepatic bile ducts; filling defects
Treatment:
- Praziquantel (single dose 40 mg/kg or 75 mg/kg in 3 divided doses): drug of choice; >95% efficacy
B. Fasciola hepatica (Sheep Liver Fluke)
- Large fluke: 3 × 1.5 cm (largest trematode to infect humans)
- Distribution: Worldwide (especially Mediterranean, South America, Africa, Asia)
- Life cycle: Definitive hosts = sheep, cattle, humans; snail intermediate host; watercress/aquatic plants as vehicles of metacercariae
- Acute phase: Young flukes migrate from gut through peritoneal cavity → liver capsule → parenchyma → bile ducts → fever, RUQ pain, hepatomegaly, eosinophilia (marked, often >3000/µL)
- Chronic phase: Adults in large bile ducts → biliary obstruction, cholangitis, bile duct fibrosis; rare cholangiocarcinoma (much less than Clonorchis)
- Eggs: Large (130-150 × 60-90 µm), yellow-brown, operculated; passed in feces
- Diagnosis: Stool microscopy; serology (ELISA - highly sensitive); imaging (liver "tracking lesions" on CT)
- Treatment: Triclabendazole (drug of choice; praziquantel NOT effective for Fasciola); nitazoxanide (alternative)
8. HEPATIC PARASITES - CESTODES (TAPEWORMS)
Echinococcus granulosus - Hydatid Cyst Disease (Sherris 8e)
Classification:
- Phylum Platyhelminthes, Class Cestoda
- Species: E. granulosus (cystic echinococcosis) and E. multilocularis (alveolar echinococcosis)
Adult Worm (Definitive Host: Dogs/Canines):
- Smallest tapeworm to cause human disease: only 3-5 mm total length
- Lives in small bowel of dogs, wolves, foxes for ~12 months
- Scolex: 4 sucking disks + double row of hooklets (armed scolex = differentiates from Taenia)
- Only 3 proglottids: 1 immature + 1 mature + 1 gravid
- Gravid proglottid splits → releases eggs (morphologically identical to Taenia eggs: hexacanth embryo inside)
Life Cycle (Herbivore-Canine Cycle; Humans as Accidental Intermediate Hosts):
- Definitive host (dog): Harbors adult worms in small bowel; passes eggs in feces
- Intermediate hosts (sheep, goats, camels, deer - normally; humans accidentally): Ingest eggs from contaminated soil/water/food
- Egg hatching: Hexacanth embryo hatches in duodenum → penetrates intestinal mucosa → enters portal blood → transported to liver (where most are trapped in sinusoids)
- Larval development: Survivors form hydatid cyst:
- Outer laminated cuticle (acellular, white, lamellar)
- Inner germinal layer (nucleated; produces brood capsules, protoscolices, daughter cysts, hydatid sand)
- Cyst fills with fluid (antigenic, anaphylactogenic)
- Growth: 1 cm over first 5-6 months → can reach >10 cm over years
- Contains "daughter cysts" (internal secondary cysts from germinal layer)
- "Hydatid sand": Degenerated protoscolices + germinal membrane fragments at bottom of cyst
- Organ distribution: Most lodge in liver (65%); next in lungs (25%); then brain, heart, bones, kidney (≤10% total)
- Cycle completion: Only when intermediate host (infected sheep) is eaten by a dog → scolices released → develop to adults
Pathogenesis:
- Slowly expanding space-occupying lesion
- Compression of adjacent liver/biliary structures
- Rupture (spontaneous or traumatic): Sudden release of antigenic cyst fluid → anaphylaxis (can be fatal); spillage of protoscolices → secondary implantation = "seeding" → multiple new cysts throughout abdomen
- Secondary bacterial infection → pyogenic liver abscess on top of hydatid cyst
Clinical Features:
- Often asymptomatic for years (slow growth)
- RUQ discomfort, hepatomegaly when cyst large
- Obstructive jaundice (biliary compression/communication)
- Rupture: Sudden intense abdominal pain + urticaria/anaphylaxis (cyst fluid = anaphylactogenic)
- Biliary communication: Cyst ruptures into bile duct → obstructive jaundice; biliary colic; bacterial superinfection; cholangitis; daughter cysts/membranes passed per rectum (pathognomonic if seen)
- Serology: ↑ serum IgE; eosinophilia (variable)
- Casoni test (intradermal): Historical; not used now
E. multilocularis (Alveolar Echinococcosis):
- Adult in foxes (not dogs); intermediate hosts = small rodents; humans accidental
- More aggressive: larval cysts invade liver as alveolar (honeycomb) structure without a defined cyst wall → infiltrates liver like a malignancy → "parasitic cancer"
- Much harder to treat; high mortality if untreated
- Distribution: Northern latitudes (Alaska, Canada, Siberia, Central Europe, China)
Diagnosis:
- Imaging (US/CT): "Water lily sign" (collapsed membranes floating in cyst fluid); daughter cysts; calcified rim; pathognomonic finding = double-line sign (two wall layers) on US
- Serological tests: ELISA for anti-Echinococcus antibodies (~85% sensitivity for hepatic cysts; lower for other organs)
- Casoni skin test: historical
- NEVER aspirate blindly → risk of spillage + anaphylaxis
- Exception: PAIR procedure (Puncture-Aspiration-Injection-Reaspiration) by expert hands with cover of albendazole
Treatment:
- Surgical resection: Traditional gold standard; requires intact cyst removal (no spillage); preceded by injection of scolicidal agent (hypertonic saline, cetrimide, ethanol)
- PAIR procedure: Minimally invasive alternative; requires albendazole cover before and after
- Albendazole (benzimidazole): Medical therapy alone for inoperable cases; also adjunct to surgery/PAIR; reduces cyst viability; given in 28-day cycles with 14-day breaks
9. HEPATIC PARASITES - PROTOZOA
A. Entamoeba histolytica - Amebic Liver Abscess
Classification: Entamoeba histolytica; Phylum Amoebozoa
Epidemiology:
- 500 million infected worldwide; 50 million develop invasive disease; 100,000 deaths/year
- Endemic in: India, Southeast Asia, Central and South America, Africa, Mexico
- Transmission: Fecal-oral (cysts in contaminated food/water)
- Most infections remain in the gut; only ~10% develop invasive disease
- Amebic liver abscess (ALA): Most common extraintestinal manifestation; more common in males (10:1) due to unknown reasons (possibly hormonal - testosterone ↑ susceptibility; estrogen protective)
Life Cycle:
- Two forms: Trophozoite (pathogenic, motile, 20-40 µm, ingests RBCs = erythrophagocytosis = hallmark) and Cyst (infective, 12-15 µm, 4 nuclei, resistant)
- Cyst ingested → trophozoites released in large intestine → colonization of colon → invasion through mucosa → trophozoites enter portal circulation → travel to liver → form abscess
Pathogenesis of ALA:
- Trophozoites produce: cysteine proteases (degrade extracellular matrix, cleave IgA/IgG), amoebapores (form pores in cell membranes → cell lysis), phospholipase A2
- Trophozoites lyse hepatocytes → liquefaction → "anchovy paste" or "chocolate sauce" pus (actually necrotic liver tissue + trophozoites; no real pus = sterile unless secondary bacterial infection)
- ALA = usually single (right lobe, 80%) large abscess; fluid is reddish-brown ("chocolate"/"anchovy paste")
- Trophozoites seen at abscess periphery (not in the necrotic center)
- Secondary bacterial superinfection: 3-5% of cases → frank pus
Clinical Features:
- Fever + RUQ pain + tender hepatomegaly = classic triad
- High fever (>39°C), sweating
- Minimal GI symptoms (only 30-40% have concurrent dysentery)
- RUQ tenderness; may have referred right shoulder pain (diaphragmatic irritation)
- Complications:
- Rupture into pleural cavity (right-sided pleural effusion, empyema): most common complication
- Rupture into pericardium (rare but life-threatening - cardiac tamponade): Left lobe ALA
- Rupture into peritoneum → peritonitis
- Rupture into bronchus → hepatobronchial fistula → patient coughs up "chocolate sauce" (pathognomonic)
Diagnosis:
- Serology (anti-amoeba IgG ELISA): >95% sensitivity for ALA; persists for years (cannot distinguish active from past infection in endemic areas)
- Aspiration of abscess: "Anchovy paste"/"chocolate sauce"; sterile; amoebic trophozoites rarely found in aspirate (at periphery)
- Stool O&P microscopy: Cysts or trophozoites; positive in only 30-40% of ALA cases
- Stool antigen test (ELISA for Gal/GalNAc lectin): More sensitive than microscopy
- Stool PCR: Most sensitive
- Imaging: Ultrasound = best initial test (round/oval hypodense lesion, right lobe); CT = confirms and characterizes
- Distinguish from pyogenic abscess: ALA = single, large; pyogenic = multiple, smaller
Treatment:
- Metronidazole (or tinidazole): 750 mg TID × 5-10 days; kills trophozoites; highly effective
- Followed by a luminal amebicide (paromomycin 25-35 mg/kg/day × 7 days; or iodoquinol): to eradicate intestinal cysts and prevent reinfection
- Aspiration: For large cysts (>10 cm), left lobe ALA (high rupture into pericardium risk), non-response to metronidazole, secondary bacterial superinfection; diagnostic aspiration to rule out pyogenic abscess
10. HEPATIC PARASITES - BLOOD FLUKES
Schistosoma - Hepatic Schistosomiasis (Sherris 8e)
Clinically relevant species:
- Schistosoma mansoni: Inferior mesenteric veins → portal and descending colon veins → portal hypertension and liver fibrosis (most common cause worldwide)
- Schistosoma japonicum: Superior mesenteric vein → small intestine and ascending colon veins → also causes severe hepatic schistosomiasis
- Schistosoma haematobium: Bladder/pelvic veins → urinary schistosomiasis (not primarily hepatic)
Adult Worm Morphology:
- Unlike other trematodes: separate sexes (males + females)
- Male: 1-2 cm; deep ventral groove (gynecophoral canal / "schist") carries female in lifelong embrace
- Female: Longer, more slender; lives inside male's groove
Life Cycle:
- Definitive hosts (humans, some mammals): Adult worms in mesenteric veins; mate → female deposits eggs (300-3000/day depending on species)
- Eggs: Secreted enzymes digest surrounding tissue; eggs near mucosa rupture into gut/bladder lumen → pass in feces/urine
- Freshwater snail (intermediate host): Eggs hatch → miracidia → penetrate snail → sporocysts → cercariae (bifurcate tail = furcocercariae)
- Cercariae in water: Actively swim; penetrate human skin on contact → lose bifurcate tail → become schistosomulae → enter dermal capillaries → systemic circulation → pulmonary capillaries → heart → aorta → portal vein → liver → mature to adults → migrate to definitive site
Egg Morphology (diagnostic):
- S. mansoni: Oval, 60 × 140 µm, prominent lateral spine
- S. japonicum: Round, 60 × 90 µm, small vestigial lateral knob (barely visible)
- S. haematobium: Oval, 115 × 65 µm, terminal spine (end spine)
Pathogenesis - Three Stages (Sherris):
-
Cercarial penetration (Swimmer's itch / Schistosome dermatitis):
- Pruritic maculopapular rash at penetration site; lasts 24-48h
- Due to hypersensitivity to cercarial antigens
-
Migration and maturation (Katayama fever):
- 4-8 weeks after infection: acute serum sickness-like syndrome
- Fever, urticaria, angioedema, marked eosinophilia, hepatosplenomegaly, lymphadenopathy
- Immune complex deposition; especially with S. japonicum (higher egg output)
- More severe with primary infections in non-immune individuals
-
Chronic egg deposition (Hepatosplenic schistosomiasis):
- Eggs not excreted get trapped in tissues → eosinophilic granuloma formation (Splendore-Hoeppli reaction) around each egg
- In the liver (portal tracts): Granulomas → fibrosis → periportal "pipestem" fibrosis (Symmers' clay-pipestem fibrosis) = pathognomonic of S. mansoni and S. japonicum
- Progressive periportal fibrosis → portal hypertension with preserved hepatocyte function (pre-sinusoidal portal hypertension)
- Liver surface: knobby, irregular
- Hepatic function is RELATIVELY PRESERVED (pre-sinusoidal block) → jaundice and hepatic encephalopathy are late findings
- Massive splenomegaly (most prominent feature; spleen may extend to pelvis)
- Esophageal varices → GI hemorrhage = major cause of death
- Secondary hypersplenism → pancytopenia
Periportal (Pipestem) Fibrosis - Key Micro Feature:
- Dense fibrous tissue deposited around portal vein branches (not around central veins)
- "Pipestem" = on cross-section, portal tracts look like pipe stems (thick fibrous wall around vessel)
- Contrast to alcoholic cirrhosis: hepatocyte architecture preserved; no nodule formation in early stages
Epidemiology:
- 200 million infected; 200,000 deaths/year → 2nd most important parasitic infection after malaria
- Distribution depends on snail host:
- S. mansoni: Africa, Nile Delta, South America, Caribbean
- S. japonicum: China, Philippines, Sulawesi
- S. haematobium: Africa, Middle East
Diagnosis:
- Stool microscopy: Characteristic eggs (S. mansoni, S. japonicum)
- Urine microscopy: S. haematobium (terminal hematuria + terminal-spined eggs)
- Stool concentration techniques (Kato-Katz method)
- Serology (ELISA): Sensitive; useful for travelers who had low egg burden; stays positive for years
- Liver biopsy: Periportal fibrosis; granulomas with eggs
- Imaging: US = periportal echogenicity ("hyperechoic ring sign"); Doppler = portal hypertension
Treatment:
- Praziquantel: Drug of choice for all Schistosoma species
- S. mansoni/haematobium: 40 mg/kg in 2 divided doses on 1 day
- S. japonicum: 60 mg/kg in 3 divided doses on 1 day
- Kills adult worms → prevents further egg deposition; some reversal of fibrosis if treated early
11. HEPATIC BACTERIA
Pyogenic Liver Abscess (PLA)
Etiology: Polymicrobial (most common) or monomicrobial:
- Gram-negatives: Escherichia coli, Klebsiella pneumoniae (monomicrobial, especially in Southeast Asia/Taiwan = "primary Klebsiella liver abscess" syndrome - also causes metastatic endophthalmitis and meningitis)
- Anaerobes: Bacteroides fragilis, other Bacteroides spp., Peptostreptococcus
- Streptococcus milleri group (S. anginosus, S. intermedius, S. constellatus): Important cause; associated with metastatic spread to brain, lung
- Staphylococcus aureus: In immunocompromised, post-surgical
- Entamoeba histolytica: Amebic abscess (technically protozoa, see above)
Sources of infection (routes to liver):
- Portal vein (pylephlebitis): Appendicitis, diverticulitis, perforated viscus, IBD, bowel malignancy with necrosis → portal bacteremia
- Biliary tract (ascending cholangitis): Biliary obstruction (stones, strictures, malignancy) → bacterial colonization → ascending infection → liver abscess; accounts for ~40% (increasing with aging population + biliary manipulation)
- Direct extension: Subphrenic abscess, perinephric abscess, perforated gallbladder
- Hematogenous (hepatic artery): Systemic bacteremia (endocarditis, IV drug use)
- Cryptogenic: No identifiable source (~20% in some series)
- Post-procedure: After percutaneous biopsy, ERCP, ablation
Klebsiella Liver Abscess (Hypervirulent K. pneumoniae):
- Predominantly in East/Southeast Asia; increasingly worldwide
- Specific hypervirulent capsular types (K1, K2) → resist phagocytosis
- Classic presentation: Primary Klebsiella liver abscess (no identifiable biliary source) in diabetics → metastatic spread to eye (endophthalmitis - often leading to blindness), CNS (meningitis, brain abscess), lungs
- hvKp (hypervirulent Klebsiella) carries hypermucoviscosity (positive string test: colony forms >5 mm string when stretched with loop)
12. HEPATITIS VIRUSES AND HCC - MOLECULAR MICROBIOLOGY SUMMARY
| Virus | Mechanism of HCC |
|---|---|
| HBV | (1) HBx protein → transcriptional activation of oncogenes (c-myc, c-fos, c-jun, Ras), inhibits p53, activates Wnt/β-catenin; (2) HBV DNA integration into host genome → insertional mutagenesis; (3) Chronic inflammation → regeneration → accumulation of driver mutations; (4) Synergy with aflatoxin B1 (p53 codon 249 G→T transversion) |
| HCV | No DNA integration (RNA virus); mechanism via chronic inflammation → cirrhosis → hepatocyte regeneration → acquisition of driver mutations; NS5A may activate PI3K/Akt; NS3 may interfere with p53; core protein inhibits apoptosis |
| Aflatoxin B1 (Aspergillus flavus/parasiticus mycotoxin; not a virus) | CYP1A2/2A3-activated AFBO (aflatoxin B1-8,9-epoxide) → adduct with guanine-N7 → specific G→T transversion at TP53 codon 249 (R249S mutation) → p53 inactivation; synergizes with HBV (>60× relative risk when both present) |
SUMMARY TABLE: HEPATIC INFECTIONS AT A GLANCE
| Organism | Type | Key Feature | Treatment |
|---|---|---|---|
| HAV | RNA virus, Picornaviridae | Fecal-oral, self-limited, never chronic | Supportive; vaccine prevention |
| HBV | DNA virus, Hepadnaviridae | Partially dsDNA, cccDNA, NTCP receptor, perinatal transmission, curable now with DAAs? (No - suppressed only) | NRTIs (tenofovir, entecavir) = suppress; pegIFN-α = functional cure |
| HCV | RNA virus, Flaviviridae | Multiple receptors (CD81, SR-B1, claudin-1), quasispecies, no cccDNA → curable | DAAs: sofosbuvir-based regimens >95% SVR |
| HDV | Defective RNA, uses HBsAg | Co-infection vs. superinfection; worst prognosis of all | Bulevirtide (NTCP entry inhibitor) + pegIFN-λ; HBV vaccination prevents HDV |
| HEV | RNA virus, Hepeviridae | Fulminant in pregnancy (HEV-1); zoonotic (HEV-3); chronic in immunocompromised | Supportive; ribavirin for chronic HEV-3; Hecolin vaccine (China) |
| E. histolytica | Protozoa | "Chocolate/anchovy paste" abscess; right lobe; erythrophagocytosis | Metronidazole → paromomycin |
| Echinococcus granulosus | Cestode larva | Hydatid cyst; dog-sheep cycle; anaphylaxis on rupture | Albendazole + PAIR/surgery |
| Clonorchis sinensis | Trematode | Chinese liver fluke; freshwater fish; cholangiocarcinoma risk | Praziquantel |
| Fasciola hepatica | Trematode | Sheep liver fluke; watercress; acute eosinophilia phase | Triclabendazole |
| Schistosoma mansoni/japonicum | Blood fluke | Pipestem periportal fibrosis; pre-sinusoidal portal hypertension; preserved hepatic function | Praziquantel |
| Klebsiella pneumoniae (hvKp) | Gram-negative bacterium | Primary liver abscess; K1/K2 capsule; diabetics; metastatic endophthalmitis | Ceftriaxone or carbapenems; drainage |
Sources: Murray Medical Microbiology 9e (9780323673228, block6 pp. 619-638), Jawetz Medical Microbiology 28e (9781260012026, block5 pp. 509-535), Sherris & Ryan Medical Microbiology 8e (9781260464283, block7 pp. 1655-1700, 1936-1950, 1941-1945)
That completes the full microbiology of the liver - covering all five hepatotropic viruses in molecular detail (structure, genome, receptors, replication, serology, disease, vaccines), hepatitis G, all major hepatic parasites (Clonorchis, Fasciola, Echinococcus, Entamoeba, Schistosoma), and bacterial liver abscess.
Remaining topics:
- Pharmacology (antivirals for HBV/HCV/HDV, drugs for portal hypertension complications, UDCA, NAC)
- Medicine / Clinical Management (Harrison's 22e)
- Surgery (Schwartz/Sabiston/Bailey & Love)
Pharma
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
This is a shared conversation. Sign in to Orris to start your own chat.