give an outline on mendelian disorders, autosomal dominant/recessive, x-linked dominant/recessive w.r.t. Robbins and Cotran pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive material from Robbins & Cotran. Let me compile the full detailed outline.
Mendelian Disorders — Outline per Robbins, Cotran & Kumar: Pathologic Basis of Disease
I. Overview: Categories of Human Genetic Disease
Genetic disorders are broadly grouped into three categories:
- Monogenic (Mendelian) Disorders — single-gene mutations with large effects; highly penetrant; follow classic Mendelian inheritance (autosomal dominant, autosomal recessive, X-linked).
- Chromosomal Disorders — structural or numerical alterations affecting autosomes or sex chromosomes; often arise de novo in the germline.
- Complex Multigenic (Multifactorial) Disorders — multiple genetic variants + environmental factors (e.g., atherosclerosis, diabetes, hypertension).
A fourth category of somatically acquired mutations (driving clonal hematopoiesis, cancers, overgrowth syndromes) is also increasingly recognized.
II. Mutations Underlying Mendelian Disorders
Mutations are permanent changes in DNA sequence. Key types:
| Type | Mechanism | Example |
|---|---|---|
| Point mutation | Single nucleotide substitution | Missense → altered amino acid; Nonsense → premature stop codon |
| Frameshift | Insertion or deletion of non-multiples of 3 bases | 4-bp insertion in HEXA → Tay-Sachs disease |
| Trinucleotide repeat expansions | Expansion of repetitive sequences | Fragile X, Huntington |
| Deletions/duplications | Larger DNA copy number changes | Affects gene dosage |
Functional Consequences of Mutations
- Loss-of-function: enzyme/transport protein deficiencies (usually AR)
- Gain-of-function: constitutively active or toxic protein products (usually AD)
Key Concepts
- Pleiotropism: one mutation → many end-organ effects (e.g., sickle cell anemia: hemolysis + vascular occlusion + splenic fibrosis + bone changes)
- Genetic heterogeneity: different gene loci → same phenotype (e.g., congenital deafness)
III. Transmission Patterns of Single-Gene Disorders
A. Autosomal Dominant (AD) Disorders
Fundamental Rules:
- Manifested in the heterozygous state
- Both males and females are equally affected and can transmit
- Affected parent → 50% chance each child is affected
- Vertical transmission through generations
Exceptions and Modifiers:
| Concept | Definition |
|---|---|
| De novo mutation | No affected parent; propensity for new mutations in germ cells of older fathers (e.g., FGFR2/FGFR3 mutations → Apert syndrome, achondroplasia) |
| Incomplete penetrance | Mutant gene carried but disease not expressed (e.g., 50% penetrance = 50% carrying the gene are affected) |
| Variable expressivity | Trait present in all carriers but at differing severity |
| Anticipation | Earlier onset and increasing severity in successive generations (e.g., trinucleotide repeat disorders) |
Biochemical Mechanisms in AD Disorders:
- Loss of regulatory proteins (e.g., tumor suppressors) — one mutant allele reduces the gene product sufficiently to alter function
- Gain-of-function — mutant protein acquires new toxic activity
- Dominant-negative effect — mutant product interferes with normal allele product (common with structural proteins like collagen and fibrillin)
Examples of AD Disorders (Table 5.1):
| System | Disorder |
|---|---|
| Neurologic | Huntington disease, neurofibromatosis, myotonic dystrophy |
| Urologic | Adult polycystic kidney disease |
| Cardiovascular | Familial hypercholesterolemia, hereditary hemorrhagic telangiectasia |
| Hematopoietic | Hereditary spherocytosis, von Willebrand disease |
| Skeletal | Marfan syndrome, Ehlers-Danlos syndrome (some variants), achondroplasia |
| Metabolic | Familial hypercholesterolemia |
Featured AD Disorder — Marfan Syndrome:
- Gene: FBN1 (fibrillin-1), chromosome 15q21
- Fibrillin-1 is an extracellular glycoprotein → component of microfibrils in connective tissue
- Mutations disrupt elastin meshworks and cause excess release of TGF-β from ECM (unopposed TGF-β signaling → tissue remodeling)
- Morphology:
- Skeletal: tall stature, dolichocephaly, arachnodactyly, hypermobile joints, pectus excavatum/carinatum, kyphoscoliosis
- Ocular: bilateral ectopia lentis (upward/outward lens subluxation) — a near-pathognomonic sign
- Cardiovascular: mitral valve prolapse (40–50%), medial degeneration of aorta → aortic dilation, aortic regurgitation, aortic dissection (most common cause of death)
B. Autosomal Recessive (AR) Disorders
Fundamental Rules:
- Disease manifests only in the homozygous state (both alleles mutated)
- Heterozygous carriers are phenotypically normal
- Both parents are typically carriers (heterozygotes)
- 25% chance of affected offspring per pregnancy; 50% carriers; 25% normal
- Often presents in siblings, not parents ("horizontal" pedigree pattern)
- More common when parents are consanguineous
Key Features Distinguishing AR from AD:
- Expression is more uniform (less variable expressivity)
- Complete penetrance is more common
- Onset often in early childhood (enzyme deficiencies manifest when both alleles are absent)
- Enzyme deficiency is the predominant mechanism — 50% residual enzyme from one functional allele is usually sufficient (hence carriers are unaffected)
Examples of AR Disorders (Table 5.2):
| System | Disorder |
|---|---|
| Metabolic | Cystic fibrosis, phenylketonuria, galactosemia, homocystinuria, lysosomal storage diseases, α₁-antitrypsin deficiency, Wilson disease, hemochromatosis, glycogen storage diseases |
| Hematopoietic | Sickle cell anemia, thalassemias |
| Endocrine | Congenital adrenal hyperplasia |
| Skeletal | Ehlers-Danlos syndrome (some variants), alkaptonuria |
| Nervous | Spinal muscular atrophy, Friedreich ataxia, neurogenic muscular atrophies |
Featured AR Disorder — Lysosomal Storage Diseases (prototype of AR enzyme deficiency):
- Inherited deficiency of lysosomal enzymes → incomplete catabolism → accumulation of insoluble intermediates within lysosomes
- Pathogenic consequences:
- Primary accumulation — engorged lysosomes interfere with cell function
- Defective autophagy — impaired mitophagy → dysfunctional mitochondria persist → free radical generation → intrinsic apoptosis
- Secondary accumulation — aggregation-prone proteins (α-synuclein, Huntingtin) accumulate
- ~70 lysosomal storage diseases identified; frequency ~1 in 5000 live births
- Treatment approaches: (1) Enzyme replacement / gene therapy; (2) Substrate reduction therapy; (3) Molecular chaperone therapy
C. X-Linked Disorders
General Principles:
- Caused by mutations in genes on the X chromosome; males are hemizygous (no corresponding Y-linked locus to compensate)
- Y chromosome male-specific region encodes few genes (mostly spermatogenesis); Y-linked mutations → male infertility → cannot be transmitted
C1. X-Linked Recessive (XLR) Disorders
Fundamental Rules:
- Males are predominantly affected (hemizygous — one mutant X is sufficient for disease)
- Females are typically carriers (heterozygous); generally unaffected because of the normal allele
- An affected male does not transmit disorder to sons (sons inherit Y chromosome from father); all daughters of an affected male are obligate carriers
- Sons of heterozygous women: 50% affected; daughters: 50% carriers
Lyon Hypothesis (X-Inactivation):
- In females, one X chromosome is randomly inactivated in each somatic cell → females are mosaics
- In most carriers, random inactivation results in ~50% cells with the normal X active → no disease
- If X-inactivation is skewed to favor inactivation of the wild-type allele → manifesting carrier female
Examples of XLR Disorders (Table 5.3):
| System | Disorder |
|---|---|
| Musculoskeletal | Duchenne muscular dystrophy |
| Hematopoietic | Hemophilia A and B, chronic granulomatous disease, G6PD deficiency |
| Immune | Agammaglobulinemia (Bruton), Wiskott-Aldrich syndrome |
| Metabolic | Diabetes insipidus (nephrogenic), Lesch-Nyhan syndrome |
| Nervous | Fragile X syndrome |
Featured XLR Disorder — G6PD Deficiency:
- X-linked; enzyme deficiency → episodic hemolytic anemia triggered by infection or oxidant drugs
- Expressed principally in males; rare manifesting carrier females result from skewed X-inactivation
C2. X-Linked Dominant (XLD) Disorders
Fundamental Rules:
- Rare; caused by dominant mutations on X chromosome
- Heterozygous females are affected (unlike XLR where females are carriers)
- Affected males tend to be more severely affected than females, or the condition may be lethal in males (e.g., incontinentia pigmenti)
- Transmission: affected mother → 50% daughters affected, 50% sons affected; affected father → all daughters affected, no sons affected
Examples:
- Fragile X syndrome (technically complex — trinucleotide expansion, discussed separately under non-classic inheritance)
- Incontinentia pigmenti (IKBKG/NEMO mutations) — lethal in males, affects females with skin, eye, neurologic, and dental abnormalities
- Rett syndrome (MECP2 mutations) — predominantly affects females
- X-linked hypophosphatemia (PHEX mutations) — rickets, affects both sexes but females less severely
IV. Comparing the Four Inheritance Patterns at a Glance
| Feature | AD | AR | XLR | XLD |
|---|---|---|---|---|
| State in which disease appears | Heterozygous | Homozygous | Hemizygous (males) | Heterozygous (females) + hemizygous (males) |
| Sex affected | Both equally | Both equally | Males >> Females | Both; females may be less severely affected |
| Carrier state | Not applicable | Heterozygotes | Heterozygous females | Not applicable (heterozygotes are affected) |
| Father → son transmission | Yes | Yes | No | No (sons get Y) |
| Risk to offspring of carrier | 50% affected | 25% affected, 50% carriers | Sons 50% affected; daughters 50% carriers | 50% children affected |
| Variable expressivity | Common | Less common | Common | Variable |
| New mutations | Common | Rare | Occasional | Occasional |
| Key molecular mechanism | Gain-of-function / dominant-negative / haploinsufficiency | Enzyme deficiency | Enzyme/protein deficiency (hemizygous) | Dominant gain- or loss-of-function on X |
V. Selected Specific Diseases Covered in Detail by Robbins & Cotran
| Disorder | Inheritance | Gene/Defect |
|---|---|---|
| Marfan syndrome | AD | FBN1 (fibrillin-1) — TGF-β excess |
| Familial hypercholesterolemia | AD | LDLR (LDL receptor) loss-of-function |
| Huntington disease | AD | HTT CAG repeat expansion |
| Neurofibromatosis type 1 | AD | NF1 (neurofibromin) |
| Cystic fibrosis | AR | CFTR — defective Cl⁻ channel |
| Phenylketonuria | AR | PAH (phenylalanine hydroxylase) deficiency |
| Sickle cell anemia | AR | HBB — Glu→Val substitution at position 6 |
| Tay-Sachs disease | AR | HEXA — 4-bp insertion → frameshift → absent hexosaminidase A |
| Gaucher disease | AR | GBA (glucocerebrosidase) deficiency |
| α₁-Antitrypsin deficiency | AR | SERPINA1 — misfolded protein accumulation in liver + uninhibited elastase in lungs |
| Duchenne muscular dystrophy | XLR | DMD (dystrophin) — frameshift deletions → absent dystrophin |
| Hemophilia A | XLR | F8 (Factor VIII) deficiency |
| G6PD deficiency | XLR | G6PD — oxidant-induced hemolysis |
| Fragile X syndrome | XLD/complex | FMR1 — CGG trinucleotide repeat expansion |
VI. Exceptions and Departures from Classic Mendelian Inheritance
Robbins & Cotran also highlights non-classic patterns that are covered in the same chapter:
- Trinucleotide repeat disorders (anticipation) — Huntington, myotonic dystrophy, Fragile X
- Genomic imprinting — Prader-Willi (paternal 15q11-q13 deletion) vs. Angelman syndrome (maternal deletion same region)
- Mitochondrial inheritance — maternally transmitted; affects organs with high energy demand
- Mosaicism — somatic or germline; explains variable expressivity
- Uniparental disomy — both copies of a chromosome derived from one parent
Source: Robbins, Cotran & Kumar — Pathologic Basis of Disease (ISBN 9780443264528), Chapter 5: Genetic Disorders, pp. 139–165
tutorial on microcytic and macrocytic anemia
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
I have all the content needed for a comprehensive tutorial. Let me now compose the full tutorial.
Tutorial: Microcytic & Macrocytic Anemias
Based on Robbins & Kumar Basic Pathology and Robbins, Cotran & Kumar Pathologic Basis of Disease
Conceptual Framework: Classifying Anemia by MCV
| MCV | Category | Key Causes |
|---|---|---|
| < 80 fL | Microcytic | Iron deficiency, thalassemia, anemia of chronic disease, sideroblastic anemia |
| 80–100 fL | Normocytic | Hemolytic anemias, aplastic anemia, acute blood loss |
| > 100 fL | Macrocytic | Megaloblastic (B12/folate deficiency), non-megaloblastic (liver disease, hypothyroidism, drugs) |
PART I — MICROCYTIC ANEMIAS (MCV < 80 fL)
The unifying pathophysiology is impaired hemoglobin synthesis, affecting heme (iron deficiency, sideroblastic), globin (thalassemia), or both.
1. Iron Deficiency Anemia (IDA)
Epidemiology: The most common nutritional deficiency worldwide. ~10% of people in high-resource countries and 25–50% in low-resource countries are anemic, with IDA the leading cause.
Iron Metabolism (Essential Background)
- Total body iron: ~3.5 g (men) / ~2.5 g (women)
- Functional pool (80%): Hemoglobin, myoglobin, iron-containing enzymes (catalase, cytochromes)
- Storage pool (15–20%): Ferritin and hemosiderin in liver macrophages, spleen, and bone marrow
| Parameter | Normal value |
|---|---|
| Serum iron | ~120 µg/dL (men), ~100 µg/dL (women) |
| Transferrin saturation | ~33% |
| TIBC | 300–350 µg/dL |
| Dietary iron (Western diet) | 10–20 mg/day |
| Daily iron loss | 1–2 mg/day (mucosal/skin cell shedding) |
Iron absorption pathway (duodenum):
- Fe³⁺ → Fe²⁺ via duodenal cytochrome B (ferric reductase)
- Fe²⁺ enters enterocyte via DMT-1 (divalent metal transporter-1)
- Fe²⁺ exits basolateral membrane via ferroportin
- Reoxidised to Fe³⁺ by hephaestin/ceruloplasmin → binds transferrin
Hepcidin: A liver-secreted peptide that negatively regulates ferroportin. Iron levels are sensed by HFE protein on hepatocytes → rising iron → rising hepcidin → less ferroportin → less absorption. Hepcidin is also upregulated by IL-6 (inflammation) and downregulated by erythroferrone (from erythroblasts during active erythropoiesis).
Causes of IDA
| Setting | Cause |
|---|---|
| High-resource countries | Chronic blood loss — GI tract (peptic ulcer, colon cancer, hemorrhoids); female genital tract (menorrhagia) |
| Low-resource countries | Inadequate intake — vegetarian diets, low bioavailability |
| Universal | Increased demand — pregnancy, infancy |
| Malabsorption | Celiac disease, gastritis, post-gastrectomy |
Stages of Iron Deficiency (in sequence)
- Depletion of iron stores → ↓ serum ferritin, absent bone marrow iron staining; no anemia yet
- Iron-limited erythropoiesis → ↓ serum iron, ↑ TIBC, ↓ transferrin saturation
- Frank IDA → microcytic hypochromic anemia, ↑ erythropoietin (marrow response blunted by iron lack)
Morphology
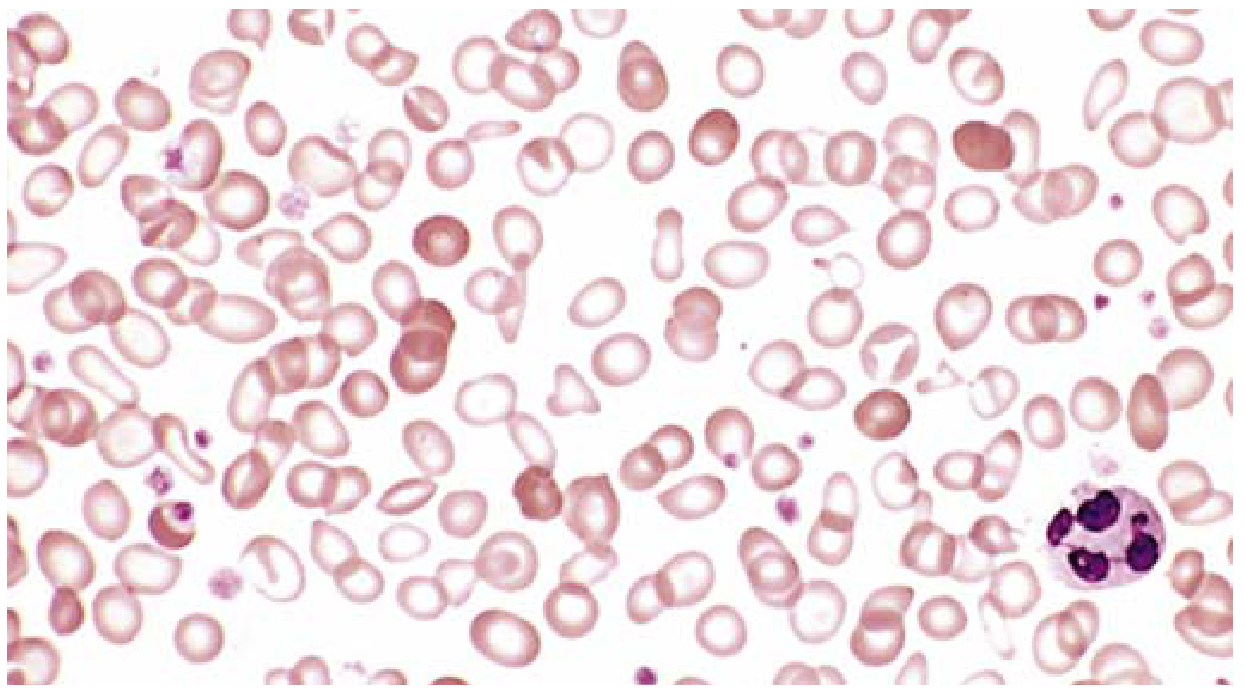
Fig. 10.10 — Iron deficiency anemia peripheral smear: microcytic, hypochromic red cells with increased central pallor. Scattered fully haemoglobinised cells from a recent transfusion appear darker. (Robbins Basic Pathology)
- Microcytic, hypochromic RBCs (MCV ↓, MCH ↓, MCHC ↓)
- Increased central pallor (>1/3 of cell diameter)
- Anisocytosis + poikilocytosis
- Platelets often elevated (reactive thrombocytosis)
- Reticulocyte count: normal or slightly low (response blunted)
Lab Findings Summary
| Test | IDA | ACD | Thalassemia trait |
|---|---|---|---|
| Serum iron | ↓ | ↓ | Normal |
| Ferritin | ↓ | ↑ | Normal/↑ |
| TIBC | ↑ | ↓ | Normal |
| Transferrin saturation | ↓ | ↓ | Normal |
| RBC count | ↓ | ↓ | ↑ or normal |
| RDW | ↑ | Normal | Normal/↑ |
Clinical Features
- Often mild and asymptomatic
- Weakness, listlessness, pallor in severe cases
- Long-standing: koilonychia (spoon nails), thin/flat nails
- Pica — compulsion to eat non-food items (dirt, clay, ice/pagophagia) — a neurobehavioral complication
- Angular cheilitis, glossitis (sore tongue)
- Impaired cognitive performance and reduced immunocompetence
"Persons often die with iron deficiency anemia but virtually never of it. Microcytic hypochromic anemia is not a disease but a symptom — always investigate the underlying cause." — Robbins Basic Pathology
2. Anemia of Chronic Disease (ACD) / Anemia of Inflammation
A functional iron deficiency — iron is abundant but sequestered and unavailable for erythropoiesis.
Common underlying conditions:
- Chronic infections: osteomyelitis, bacterial endocarditis, lung abscess
- Chronic immune disorders: rheumatoid arthritis, Crohn disease
- Cancers: Hodgkin lymphoma, lung/breast carcinoma
Pathogenesis: Pro-inflammatory cytokines (especially IL-6) → ↑ hepatic hepcidin → hepcidin downregulates ferroportin on marrow macrophages → iron is trapped in macrophages and cannot be delivered to erythroblasts. Additionally, chronic inflammation blunts renal erythropoietin synthesis.
Key distinguishing lab feature:
- Serum iron: ↓ (same as IDA)
- Ferritin: ↑ (iron sequestered, not depleted) — key differentiator
- TIBC: ↓ (unlike IDA where TIBC is ↑)
- Red cells: mildly hypochromic and microcytic or normocytic
Treatment: Treat the underlying condition; erythropoietin + iron can temporarily improve anemia.
3. Thalassemias
Definition: Inherited disorders of globin chain synthesis causing reduced (or absent) production of α- or β-globin chains.
Genetics:
- β-globin gene: single gene on chromosome 11 (mutations = mainly point mutations affecting transcription, splicing, or translation of β-globin mRNA)
- α-globin genes: two tandem genes on chromosome 16 per haploid genome (4 total; mutations = mainly gene deletions)
- Autosomal codominant inheritance
Pathogenesis: Reduced globin synthesis → (1) hemoglobin deficiency → microcytic hypochromic anemia; (2) excess unpaired globin chains precipitate → intracellular inclusions → RBC membrane damage → hemolysis and ineffective erythropoiesis.
β-Thalassemia
| Syndrome | Genotype | Clinical Features |
|---|---|---|
| β-Thalassemia major (Cooley anemia) | β⁰/β⁰ (no β-chain) | Severe transfusion-dependent anemia; splenomegaly; growth retardation; extramedullary hematopoiesis; facial bone changes ("chipmunk face"); iron overload |
| β-Thalassemia intermedia | β⁺/β⁰ or β⁺/β⁺ | Moderately severe; transfusions not required |
| β-Thalassemia minor (trait) | β⁺/β (one normal allele) | Asymptomatic; mild/absent anemia; ↑ HbA2; often mistaken for IDA — MCV low but RBC count high |
α-Thalassemia
| Syndrome | Gene deletions | Clinical Features |
|---|---|---|
| Silent carrier | 1 deleted (−/α, α/α) | No abnormality; asymptomatic |
| α-Thalassemia trait | 2 deleted | Asymptomatic; resembles β-thal minor |
| HbH disease | 3 deleted (−/−, −/α) | Moderate anemia (resembles β-thal intermedia); HbH (β₄ tetramers) |
| Hydrops fetalis | 4 deleted (−/−, −/−) | Lethal in utero; Hb Bart's (γ₄ tetramers); incompatible with extrauterine life |
Key point: Thalassemia trait is commonly misdiagnosed as IDA — distinguish by: normal/↑ ferritin, normal/↑ TIBC, ↑ RBC count, ↑ HbA2 on HPLC (in β-thal minor).
4. Sideroblastic Anemia
Defining lesion: Ringed sideroblasts — abnormal erythroid precursors in which iron-laden mitochondria form a perinuclear ring (seen on Prussian blue stain of bone marrow).
Mechanism: Disruption of heme synthesis → iron cannot be incorporated into protoporphyrin → accumulates in mitochondria around the nucleus.
Types:
| Form | Cause |
|---|---|
| Inherited (X-linked) | Mutations in ALAS2 gene (ALA synthase 2 — first step of heme synthesis) |
| Inherited (AR) | Mutations in SLC25A38 (glycine importer) |
| Acquired — MDS | Myelodysplastic syndrome (most common acquired form) |
| Acquired — drugs/toxins | Ethanol, isoniazid, pyrazinamide, linezolid |
| Acquired — nutritional | Copper deficiency (also zinc excess) |
Clinical: Microcytic anemia in inherited forms; dimorphic RBC population (microcytic + normocytic/macrocytic mix) in acquired forms. Copper deficiency also causes myelopathy.
Treatment: Pyridoxine (vitamin B6) for ALAS2 mutations (some respond); discontinue offending drug for acquired forms; treat underlying MDS.
PART II — MACROCYTIC ANEMIAS (MCV > 100 fL)
Divided into megaloblastic and non-megaloblastic types.
Megaloblastic Anemias
Common theme: Impaired DNA synthesis → nuclear-cytoplasmic asynchrony → ineffective hematopoiesis.
Pathogenesis: Vitamin B12 and folate are required for synthesis of thymidine (one of the four DNA bases). Deficiency → defective DNA replication → rapidly dividing cells most affected (marrow, GI epithelium). Two consequences:
- Many progenitors trigger DNA damage response → apoptosis (ineffective erythropoiesis)
- Surviving progenitors produce fewer, larger red cells (fewer cell divisions → larger cells)
Universal Morphologic Features of Megaloblastic Anemia
- Macro-ovalocytes (large, oval RBCs without central pallor — hyperchromic appearance, but MCHC is not truly elevated)
- Marked anisocytosis and poikilocytosis
- Hypersegmented neutrophils (5+ lobes in a single neutrophil, or ≥1 neutrophil with 6+ lobes) — pathognomonic
- Low reticulocyte count
- Hypercellular bone marrow with megaloblastic changes: giant bands, giant metamyelocytes, large erythroid precursors with immature-appearing ("open") nuclei relative to mature cytoplasm
5. Folate Deficiency Anemia
Sources of folate: Green leafy vegetables, liver, dairy. Heat-labile (destroyed by cooking).
Body stores: Only 5–20 mg total; sufficient for only 3–4 months — deficiency develops quickly.
Causes of folate deficiency:
| Category | Examples |
|---|---|
| Decreased intake | Poor diet, alcoholism (most common in high-resource countries), infancy |
| Impaired absorption | Malabsorption, intrinsic intestinal disease, anticonvulsants, oral contraceptives |
| Increased loss | Hemodialysis |
| Increased requirement | Pregnancy, infancy, disseminated cancer, markedly increased hematopoiesis |
| Impaired utilization | Folate antagonists (methotrexate, trimethoprim) |
Clinical features:
- Megaloblastic anemia (identical hematology to B12 deficiency)
- GI mucosal changes: sore tongue, glossitis
- NO neurologic manifestations (key distinguishing feature from B12 deficiency)
Diagnosis: ↓ serum folate, ↓ RBC folate, ↑ serum homocysteine, normal methylmalonate (distinguishes from B12 deficiency).
Critical: Folate supplementation corrects the anemia of B12 deficiency but does NOT prevent — and may worsen — the neurologic damage. Always exclude B12 deficiency before starting folate therapy.
6. Vitamin B12 (Cobalamin) Deficiency Anemia
Sources: Animal products (meat, fish, dairy, eggs). Heat-stable. Also synthesised by gut flora.
Body stores: Liver stores 2–5 mg — sufficient for 5–20 years. Clinical presentation therefore follows years of unrecognised malabsorption.
Absorption pathway:
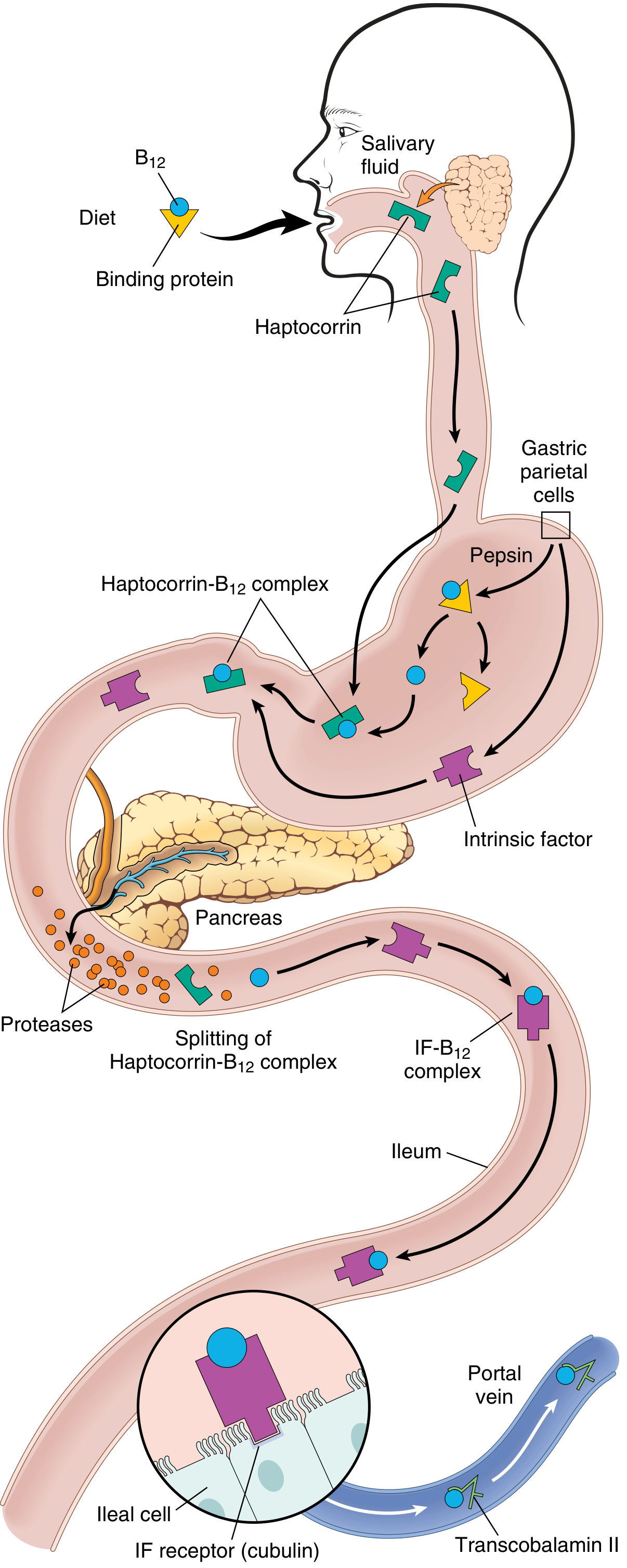
Fig. 10.12 — Vitamin B12 absorption: dietary B12 → stomach (freed by pepsin, binds haptocorrin) → duodenum (pancreatic proteases release B12, binds intrinsic factor) → terminal ileum (IF-B12 complex binds cubilin receptor on ileal enterocytes) → absorbed, bound to transcobalamin II → delivered to liver and bone marrow. (Robbins Basic Pathology)
Causes of B12 Deficiency
| Cause | Mechanism |
|---|---|
| Pernicious anemia (most common) | Autoimmune atrophic gastritis → loss of parietal cells → absent intrinsic factor. Serum autoantibodies to IF (diagnostic but not primary pathogen). |
| Gastrectomy | Loss of IF-producing cells |
| Ileal resection / Crohn disease / Whipple disease | Loss of IF-B12 absorbing cells |
| Blind loop / diverticulosis | Bacterial overgrowth → competitive uptake |
| Fish tapeworm (Diphyllobothrium) | Competitive parasitic uptake |
| Gastric atrophy / achlorhydria | Cannot release B12 from food-bound form (especially elderly) |
| Strict veganism | Only cause of dietary B12 deficiency |
Why B12 Deficiency Causes Neurologic Damage (and Folate Deficiency Does Not)
Vitamin B12 has two unique metabolic roles:
- Methylation of homocysteine → methionine (requires methylcobalamin; regenerates tetrahydrofolate → thymidine synthesis)
- Isomerisation of methylmalonyl-CoA → succinyl-CoA (requires adenosylcobalamin)
Folate deficiency only affects role #1 (thymidine synthesis). B12 deficiency affects both. Defective methylmalonyl-CoA conversion accumulates methylmalonic acid — which disrupts myelin synthesis in neuronal cells.
Neurologic lesion — Subacute Combined Degeneration (SCD):
- Demyelination of posterior columns (dorsal) — loss of vibration sense, proprioception
- Demyelination of lateral columns (corticospinal tracts) — spastic weakness, hyperreflexia
- Peripheral neuropathy — symmetric tingling, numbness, burning in feet/hands
- Neurologic damage may be irreversible even after B12 treatment
Clinical Features of Pernicious Anemia
| Feature | Detail |
|---|---|
| Anemia | Pallor, fatigue, dyspnea, palpitations |
| Mild jaundice | Ineffective erythropoiesis → intramedullar haemolysis |
| Glossitis | "Beefy red tongue" — megaloblastic changes in oral mucosa |
| Neurologic | SCD: symmetric paraesthesias → unsteady gait → loss of position sense |
| Gastric | Autoimmune atrophic gastritis; increased risk of gastric carcinoma |
Diagnosis of B12 Deficiency
| Finding | Result |
|---|---|
| Serum B12 | ↓ |
| Serum folate | Normal or ↑ |
| Serum homocysteine | ↑ |
| Serum methylmalonate | ↑ (unique to B12 deficiency) |
| Blood smear | Macro-ovalocytes, hypersegmented neutrophils |
| Bone marrow | Hypercellular, megaloblastic changes |
| Anti-intrinsic factor antibodies | Present in pernicious anemia (specific) |
Response to treatment: Reticulocytosis in 2–3 days after parenteral B12; anemia resolves over weeks. Neurologic deficits often persist or are only partially reversible.
Differentiating B12 from Folate Deficiency
| Feature | B12 Deficiency | Folate Deficiency |
|---|---|---|
| Serum B12 | ↓ | Normal |
| Serum folate | Normal or ↑ | ↓ |
| RBC folate | Normal | ↓ |
| Homocysteine | ↑ | ↑ |
| Methylmalonate | ↑ | Normal |
| Neurologic deficits | YES (SCD) | NO |
| Onset | Slow (years) | Faster (months) |
Non-Megaloblastic Macrocytosis
Macrocytic anemia without hypersegmented neutrophils or megaloblastic marrow changes. Mechanisms differ:
| Cause | Mechanism |
|---|---|
| Liver disease / alcoholism | Lipid membrane abnormalities → ↑ RBC membrane surface area → macrocytes (also folate deficiency component) |
| Hypothyroidism | Slowed RBC maturation |
| Drugs (hydroxyurea, AZT, azathioprine) | Inhibit DNA synthesis but without classic megaloblastic morphology |
| Reticulocytosis | Reticulocytes are larger → MCV rises after hemolysis/hemorrhage treatment |
| MDS | Dyserythropoiesis |
Master Summary Table
| Feature | IDA | ACD | Thalassemia Minor | Sideroblastic | Folate Def | B12 Def |
|---|---|---|---|---|---|---|
| MCV | ↓ | Normal/↓ | ↓ | ↓ (inherited); variable (acquired) | ↑ | ↑ |
| Serum iron | ↓ | ↓ | Normal | ↑ | Normal | Normal |
| TIBC | ↑ | ↓ | Normal | Normal | Normal | Normal |
| Ferritin | ↓ | ↑ | Normal/↑ | ↑ | Normal | Normal |
| Transferrin sat | ↓ | ↓ | Normal | ↑ | Normal | Normal |
| Homocysteine | — | — | — | — | ↑ | ↑ |
| Methylmalonate | — | — | — | — | Normal | ↑ |
| Smear hallmark | Microcytic, hypochromic | Mild hypo/normo | Target cells, microcytic | Dimorphic, ring sideroblasts on BM | Macro-ovalocytes, hyperseg PMN | Macro-ovalocytes, hyperseg PMN |
| Neuro deficits | No | No | No | No (unless Cu deficiency) | No | YES (SCD) |
| BM finding | ↑ cellularity, no iron stores | ↑ iron in macrophages | Hyperplastic erythropoiesis | Ringed sideroblasts | Megaloblastic | Megaloblastic |
Sources: Robbins & Kumar Basic Pathology (ISBN 9780323790185), Ch. 10, pp. 393–397; Robbins, Cotran & Kumar Pathologic Basis of Disease (ISBN 9780443264528), Ch. 14, pp. 608–616; Goldman-Cecil Medicine, Ch. 145.
topographic anatomy of upper limb with nomenclature, and osteology of clavicle in easy language
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content from Gray's Anatomy for Students and Imaging Anatomy. Let me compile the full tutorial.
Topographic Anatomy of the Upper Limb + Osteology of the Clavicle
Based on Gray's Anatomy for Students and Imaging Anatomy: Bones, Joints, Vessels and Nerves
PART I — TOPOGRAPHIC ANATOMY OF THE UPPER LIMB
What is "topographic anatomy"?
Topographic (regional) anatomy means studying the body by area/region rather than by system. Instead of studying "all muscles" or "all nerves," you look at everything — bones, muscles, nerves, vessels — within one region at a time. For the upper limb, these regions are stacked from top to bottom.
1. Overview: The Upper Limb as a Whole
The upper limb is a jointed tool whose main job is to place the hand in a useful position. Every structure from the shoulder down to the fingertips serves this goal.
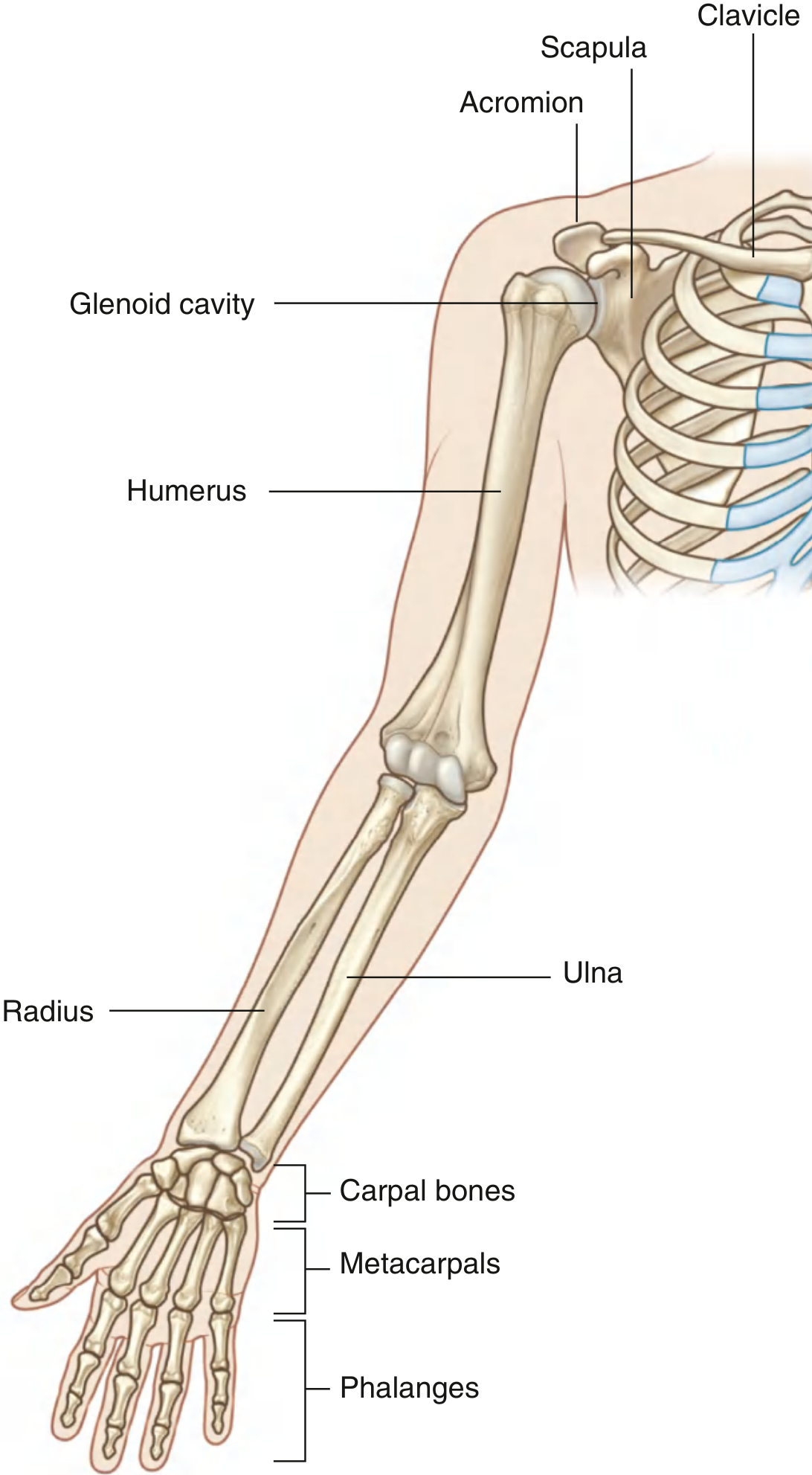
Fig. 7.7 — Bones of the shoulder and entire upper limb (Gray's Anatomy for Students)
2. Nomenclature: Names of Regions
The upper limb is divided into named regions. Each region has its own official anatomical name:
| Common name | Anatomical/Technical name |
|---|---|
| Shoulder | Regio deltoidea (deltoid region) + Regio scapularis |
| Armpit | Axilla (or axillary fossa) |
| Upper arm | Brachium (→ adjective: brachial) |
| Front of elbow | Fossa cubitalis (cubital fossa) |
| Forearm | Antebrachium (→ adjective: antebrachial) |
| Wrist | Carpus (→ adjective: carpal) |
| Palm | Palma manus |
| Back of hand | Dorsum manus |
| Fingers | Digiti manus (digits of the hand) |
| Thumb | Pollex (digit I) |
| Index finger | Index (digit II) |
| Middle finger | Digitus medius (digit III) |
| Ring finger | Digitus anularis (digit IV) |
| Little finger | Digitus minimus / minimus manus (digit V) |
| Finger bones | Phalanges (singular: phalanx) |
3. The Bones and Their Joints — Region by Region
A. Shoulder (Regio Scapularis / Deltoidea)
Three bones form the shoulder:
- Clavicle ("collar bone") — horizontal strut connecting the trunk to the upper limb
- Scapula ("shoulder blade") — flat triangular bone sitting on the back of the rib cage
- Humerus — the single bone of the upper arm; its upper end (head) sits in the socket of the scapula
Joints of the shoulder:
| Joint | Bones involved | Movement |
|---|---|---|
| Sternoclavicular (SC) joint | Clavicle + manubrium of sternum | Only bony attachment of upper limb to trunk |
| Acromioclavicular (AC) joint | Clavicle + acromion of scapula | Gliding; transmits forces from limb to clavicle |
| Glenohumeral joint | Head of humerus + glenoid cavity of scapula | Ball-and-socket; most mobile joint in the body |
Movements at the glenohumeral joint:
- Flexion (arm forward), Extension (arm backward)
- Abduction (arm away from body), Adduction (arm toward body)
- Medial rotation (internal rotation), Lateral rotation (external rotation)
- Circumduction (combination of all the above in a circle)
B. Axilla (Armpit)
The axilla is the gateway to the upper limb — a pyramidal space where all the major nerves, arteries, and veins travelling between the neck and the arm pass through.
Boundaries of the axilla:
| Wall | What forms it |
|---|---|
| Anterior wall | Pectoralis major + pectoralis minor muscles |
| Posterior wall | Subscapularis, teres major, latissimus dorsi |
| Medial wall | Serratus anterior muscle on ribs 1–4 |
| Lateral wall | Intertubercular groove of the humerus (narrowest wall) |
| Apex (inlet) | Clavicle (front) + superior border of scapula (back) + 1st rib (medial) |
| Base (floor) | Axillary fascia + skin of the armpit |
Contents of the axilla (the important structures passing through):
- Axillary artery (continuation of subclavian artery — divided into 3 parts by the pectoralis minor)
- Axillary vein (formed by union of basilic vein + brachial veins)
- Brachial plexus (the main nerve network of the upper limb — formed from C5 to T1)
- Axillary lymph nodes (drain the breast, arm, and thoracic wall — clinically important in breast cancer)
C. Arm (Brachium)
One bone: the humerus
The arm has two muscle compartments separated by medial and lateral intermuscular septa attached to the humerus:
| Compartment | Position | Main muscles | Main action |
|---|---|---|---|
| Anterior compartment | Front of arm | Biceps brachii, brachialis, coracobrachialis | Flexion of forearm at elbow |
| Posterior compartment | Back of arm | Triceps brachii | Extension of forearm at elbow |
Key nerves of the arm:
- Musculocutaneous nerve (C5, C6) — supplies anterior compartment; becomes the lateral cutaneous nerve of the forearm
- Radial nerve (C5–C8, T1) — runs in the spiral groove of the humerus; supplies posterior compartment
- Median nerve (C6–C8, T1) — runs in front of the arm; no branches in arm
- Ulnar nerve (C8, T1) — passes behind the medial epicondyle ("funny bone")
D. Elbow Joint + Cubital Fossa
Elbow joint: hinge joint between the distal humerus and the proximal radius + ulna.
- Flexion and extension of the forearm
- Also allows the radius to spin on the capitulum (part of humeral condyle) during pronation/supination
Cubital fossa = the triangular hollow at the front of the elbow. Contents (lateral to medial: TAN):
- Tendon of biceps
- Artery (brachial artery — divides here into radial and ulnar)
- Nerve (median nerve)
E. Forearm (Antebrachium)
Two bones:
- Radius — lateral bone (thumb side); head at top, wide lower end forming most of the wrist joint
- Ulna — medial bone (little finger side); large olecranon process at top ("elbow tip")
The radius and ulna are joined by the interosseous membrane and can rotate relative to each other — this allows pronation and supination.
| Movement | What happens | Result |
|---|---|---|
| Supination | Radius parallel to ulna | Palm faces forward (anatomical position) |
| Pronation | Radius crosses over ulna | Palm faces backward |
Two compartments of the forearm:
| Compartment | Position | Action |
|---|---|---|
| Anterior (flexor) | Front | Flexion of wrist and fingers; pronation |
| Posterior (extensor) | Back | Extension of wrist and fingers; supination |
F. Wrist (Carpus) and Hand
Wrist joint: formed between the distal radius (and articular disc distal to the ulna) and the proximal row of carpal bones.
Movements at the wrist: flexion, extension, abduction (radial deviation), adduction (ulnar deviation), circumduction.
Carpal bones (8 total) — in two rows:
Proximal row (lateral to medial): Scaphoid, Lunate, Triquetrum, Pisiform
Distal row (lateral to medial): Trapezium, Trapezoid, Capitate, Hamate
Easy mnemonic: "Some Lovers Try Positions That They Can't Handle"
Metacarpals (5): form the skeleton of the palm. Metacarpal I belongs to the thumb, which has a special saddle joint (carpometacarpal joint I) allowing greater freedom of movement — including the key movement of opposition (touching thumb to fingertips).
Phalanges: finger bones
- Thumb: 2 phalanges (proximal + distal)
- Other 4 fingers: 3 phalanges each (proximal + middle + distal)
Hand joints:
- Metacarpophalangeal (MCP) joints — knuckles; biaxial, allow flexion/extension + abduction/adduction
- Proximal interphalangeal (PIP) joints — hinge joints
- Distal interphalangeal (DIP) joints — hinge joints
4. Muscles: General Compartment Logic
| Region | Anterior compartment | Posterior compartment |
|---|---|---|
| Arm | Flexors (biceps, brachialis) | Extensors (triceps) |
| Forearm | Flexors of wrist + fingers; pronators | Extensors of wrist + fingers; supinator |
| Hand | Intrinsic muscles (thenar, hypothenar, lumbricals, interossei) | — |
Shoulder muscles wrap around the glenohumeral joint:
- Rotator cuff (4 muscles — SITS): Supraspinatus, Infraspinatus, Teres minor, Subscapularis — form a cuff around the joint and stabilize the humeral head in the glenoid
5. Innervation — Brachial Plexus in Brief
All muscles and skin of the upper limb (below the shoulder girdle) are innervated by the brachial plexus, formed from spinal roots C5, C6, C7, C8, and T1.
Clinical testing of spinal levels in the upper limb:
| Spinal level | Test movement (Myotome) | Test area (Dermatome) |
|---|---|---|
| C5 | Arm abduction at shoulder | Upper lateral arm |
| C6 | Forearm flexion at elbow | Palmar thumb |
| C7 | Forearm extension at elbow | Pad of index finger |
| C8 | Finger flexion | Pad of little finger |
| T1 | Finger abduction/adduction | Medial elbow skin |
Tendon reflexes:
- Biceps reflex → tests C6
- Triceps reflex → tests C7
Four terminal nerves and what they supply:
| Nerve | Spinal roots | Main territory |
|---|---|---|
| Musculocutaneous | C5, C6 | Anterior arm (biceps, brachialis) |
| Radial | C5–C8, T1 | All extensors; dorsal hand skin |
| Median | C6–C8, T1 | Anterior forearm most flexors; lateral palm + 3½ fingers |
| Ulnar | C8, T1 | Intrinsic hand muscles; medial 1½ fingers |
6. Blood Supply
- Subclavian artery → becomes axillary artery (at lateral border of 1st rib) → becomes brachial artery (at lower border of teres major)
- Brachial artery divides at the cubital fossa into radial artery (lateral) and ulnar artery (medial)
- Radial + ulnar arteries anastomose in the hand to form the superficial and deep palmar arches, which supply the digits
Superficial veins (clinically important for IV access):
- Cephalic vein — runs on lateral (radial) side of forearm and arm; drains into axillary vein
- Basilic vein — runs on medial (ulnar) side; pierces deep fascia of arm, joins brachial vein → forms axillary vein
- Median cubital vein — connects cephalic to basilic across the cubital fossa; most common site for venepuncture
7. Relationship to the Neck
The upper limb is directly connected to the neck through the axillary inlet — a triangular opening bounded by:
- Rib I (medially)
- Posterior surface of clavicle (anteriorly)
- Superior border of scapula (posteriorly)
- Coracoid process (laterally, forming the apex)
All vessels and nerves passing between the neck and upper limb cross over rib I through this inlet.
PART II — OSTEOLOGY OF THE CLAVICLE
(Explained in simple language)
What is the Clavicle?
The clavicle (from Latin clavicula = "little key") is your collar bone — the horizontal strut you can feel just below your neck on each side. It is the only bony link between your arm and your trunk. Without it, your shoulder would collapse inward.
Shape
The clavicle has a gentle S-shape when viewed from above:
- The medial (inner) two-thirds curves forward (convex anteriorly) — like the front of the letter "S"
- The lateral (outer) one-third curves backward (concave anteriorly)
Think of it as a flattened, twisted rod. This S-curve gives it mechanical strength and flexibility.
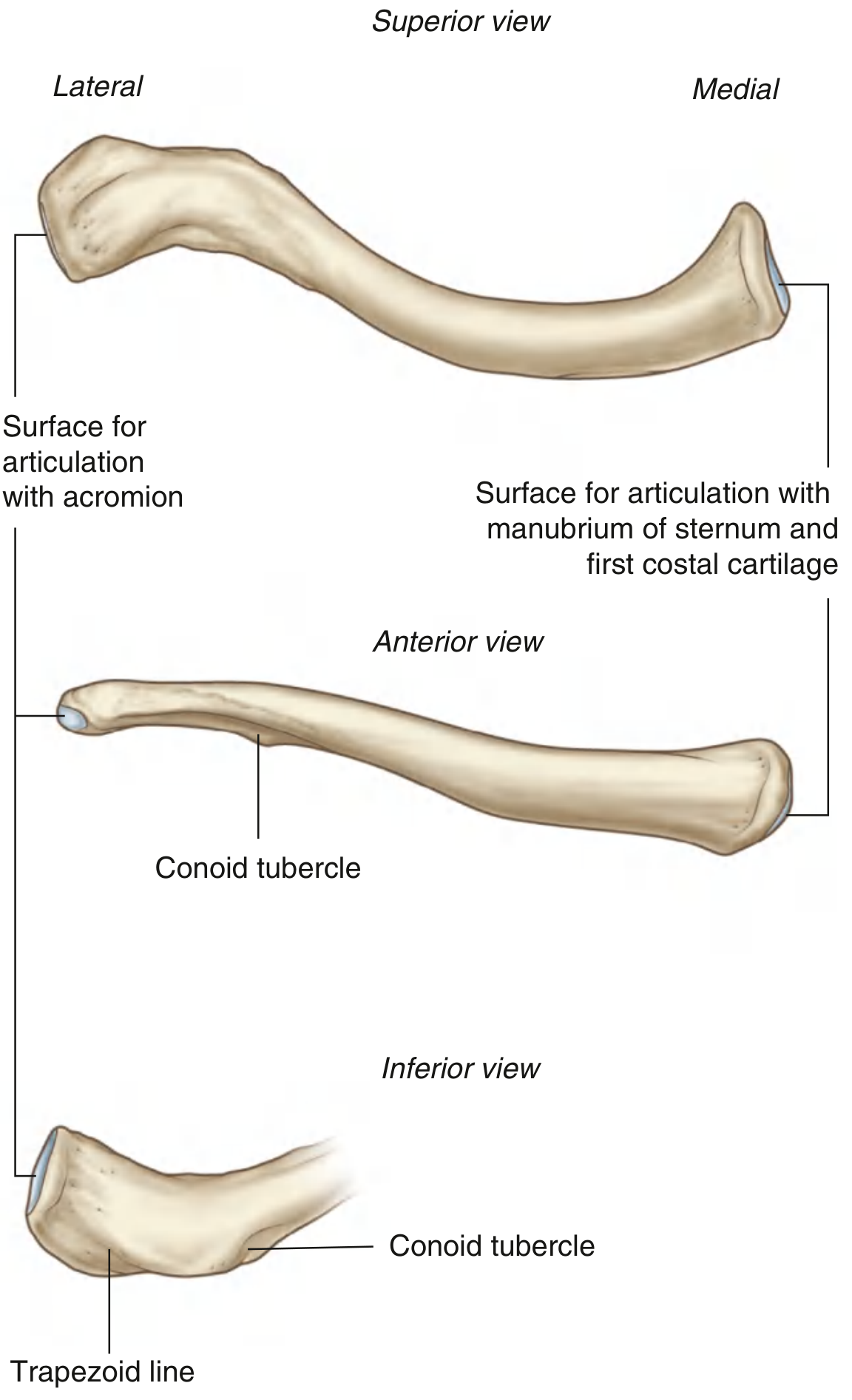
Fig. 7.20 — Right clavicle (Gray's Anatomy for Students): superior view (top), anterior view (middle), inferior view (bottom)
Parts of the Clavicle
The clavicle has two ends and a shaft (body) connecting them:
1. Sternal End (Medial End)
- Shape: Rounded, quadrangular/bulky — the bigger, heavier end
- Has a large articular facet that joins the manubrium of the sternum (breastbone) and partially the 1st costal cartilage → forms the sternoclavicular (SC) joint
- This is the only true bony joint between the upper limb and the axial skeleton (trunk)
- The inferior surface here has an oval depression = impression for the costoclavicular ligament (the strong ligament anchoring the clavicle to rib 1)
2. Acromial End (Lateral End)
- Shape: Flat and broad — thinner, flattened
- Has a small oval facet on its tip that joints with a matching facet on the acromion of the scapula → forms the acromioclavicular (AC) joint
- This joint is felt as a small bump on top of the shoulder
3. Shaft (Body)
- The middle portion connecting the two ends
- The superior surface is relatively smooth — no major muscle attachments here
- The inferior surface is rougher and carries important markings (see below)
Key Bony Markings (What you find on the surface)
| Marking | Location | What attaches |
|---|---|---|
| Conoid tubercle | Inferior surface, junction of lateral 1/3 and middle 1/3 | Conoid part of coracoclavicular ligament |
| Trapezoid line | Inferior surface, lateral to conoid tubercle, running forward/laterally | Trapezoid part of coracoclavicular ligament |
| Impression for costoclavicular ligament | Inferior surface near sternal end | Costoclavicular ligament (holds clavicle down to rib 1) |
| Deltoid tubercle | Anterior surface, lateral half | Deltoid muscle (origin) |
| Trapezoid tubercle | Posterior surface, lateral half | Trapezius muscle (insertion) |
Coracoclavicular ligament = the main suspensory ligament that holds the scapula (and the whole arm) up from the clavicle. It has two parts: the conoid (medial, cone-shaped) and the trapezoid (lateral, flat).
Muscle Attachments on the Clavicle
The clavicle acts as an anchor for several muscles connecting the arm to the trunk and neck:
| Region | Surface | Muscle | What it does |
|---|---|---|---|
| Medial half anterior | Anterior | Pectoralis major (clavicular head) | Flex + medially rotate arm |
| Medial half posterior | Posterior | Sternohyoid | Depresses hyoid bone |
| Medial half superior | Superior | Sternocleidomastoid | Turns head to opposite side |
| Lateral half anterior | Anterior | Deltoid | Abducts arm |
| Lateral half posterior | Posterior | Trapezius | Elevates scapula |
Ossification (How the Clavicle Develops) — Simple Explanation
The clavicle is special among all bones in the body in two ways:
-
First bone to start forming in the fetus — ossification begins around the 5th week of pregnancy, before any other bone in the body.
-
Last bone to completely fuse — the epiphysis (growth plate) at the sternal end does not fuse until 22–25 years of age — making it the last epiphysis in the entire body to close. This is clinically important: an injury to the medial end of the clavicle in a young adult may be a physeal (growth plate) injury rather than a true joint dislocation.
How it ossifies:
- Most of the clavicle ossifies by intramembranous (desmal) ossification — directly from connective tissue, without a cartilage model first (unlike most other long bones)
- The sternal end develops from a secondary ossification centre and fuses by endochondral ossification
- The acromial end ossifies by intramembranous ossification
Clinical Relevance of the Clavicle
| Clinical point | Explanation |
|---|---|
| Most commonly fractured bone in the body | Falls on outstretched hand (FOOSH) transmit force to the clavicle; it typically breaks at the junction of middle and lateral thirds (weakest point, no muscular reinforcement) |
| Fracture causes | The medial fragment is pulled up by sternocleidomastoid; the lateral fragment drops down and inward due to the weight of the arm |
| Why the shape matters | The S-curve makes the middle third weak but also ensures that a fracture here (rather than at the ends) protects the underlying subclavian vessels and brachial plexus |
| AC joint injury ("shoulder separation") | Disruption of the acromioclavicular joint + coracoclavicular ligaments; the shoulder drops away from the clavicle |
| SC joint dislocation | Much less common; a posterior dislocation is dangerous because the sternal end can compress the trachea, esophagus, or great vessels |
| Nutrient canal | An oblique nutrient canal runs in the shaft directed toward the acromial end; in ~6–10% of people there is also an accessory canal for the supraclavicular nerve |
Summary: Clavicle at a Glance
| Feature | Detail |
|---|---|
| Type | Long bone (S-shaped) |
| Classification | Part of the shoulder girdle (pectoral girdle) |
| Articulations | Medially: manubrium + 1st costal cartilage (SC joint); laterally: acromion (AC joint) |
| Function | Only bony connection between upper limb and trunk; strut to keep shoulder wide |
| First bone to ossify | 5th week of fetal life |
| Last epiphysis to fuse | Sternal end, at 22–25 years |
| Most common fracture site | Junction of middle + lateral thirds |
| Key ligaments attached | Costoclavicular, coracoclavicular (conoid + trapezoid) |
Sources: Gray's Anatomy for Students (ISBN 9780323934237), Chapter 7 — Upper Limb; Imaging Anatomy: Bones, Joints, Vessels and Nerves, Vol. 3 (ISBN 9781626239845)
Create a visual anatomy poster of upper limb regions and landmarks
Generating Image
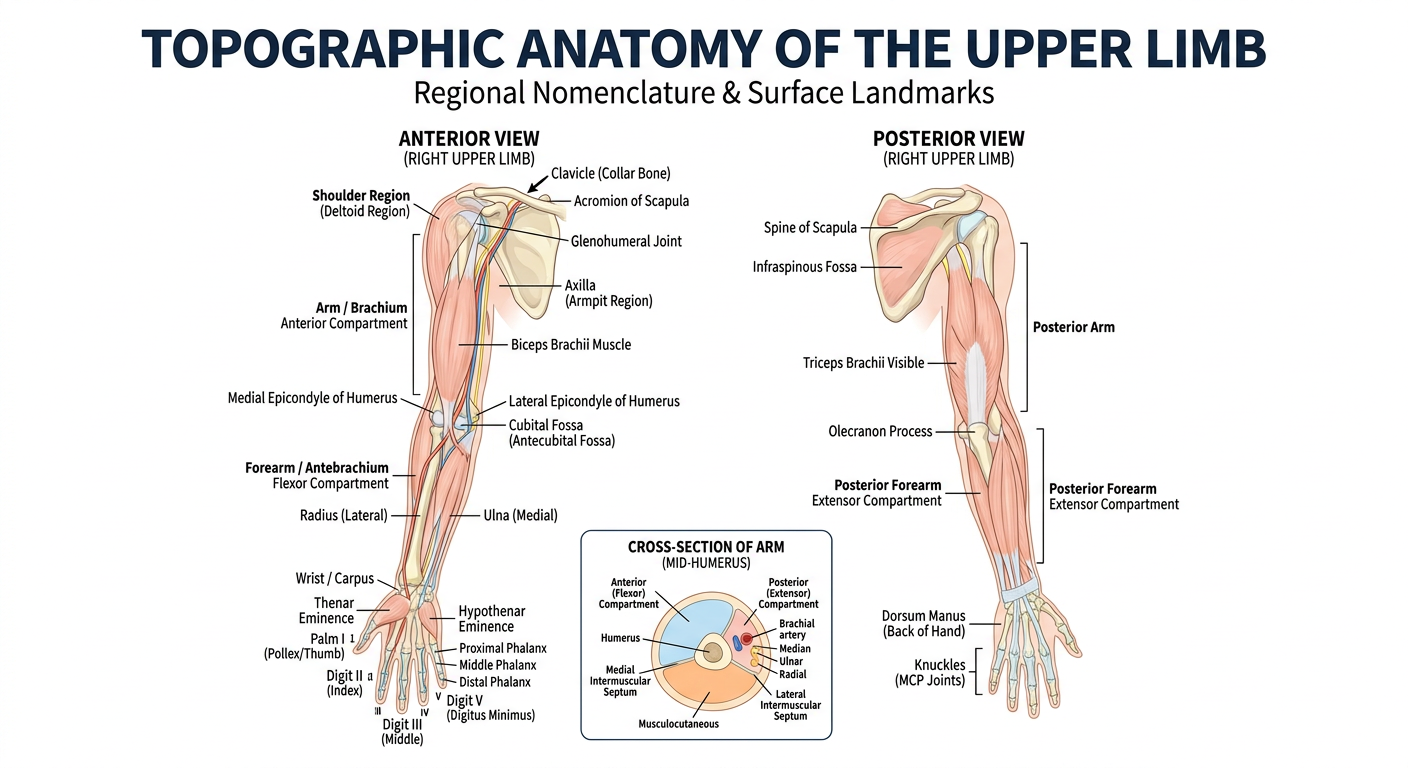
Here is your anatomy poster covering the key topographic regions and surface landmarks of the upper limb. It includes:
Anterior (left) view:
- Shoulder girdle — clavicle, acromion, glenohumeral joint
- Axilla (armpit gateway)
- Brachium — biceps brachii, medial/lateral epicondyles
- Cubital fossa (antecubital fossa)
- Antebrachium — radius (lateral) and ulna (medial)
- Carpus, thenar/hypothenar eminences, palma manus
- Digits I–V with phalangeal labeling
Posterior (right) view:
- Spine of scapula, infraspinous fossa
- Triceps brachii, olecranon process
- Extensor compartment of forearm
- Dorsum manus and MCP knuckles
Bottom inset: Cross-sectional view of the arm showing the anterior flexor compartment (blue) and posterior extensor compartment (orange) separated by intermuscular septa around the humerus.
upper limb w.r.t. KLM oriented anatomy whole, in easy language
Asking for Preferences
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Note: Moore's Clinically Oriented Anatomy isn't in the library by that exact title. The closest available equivalent is Gray's Anatomy for Students, which follows the same regional, clinically oriented approach — organized region by region with clinical boxes, just like Moore. The following tutorial is fully drawn from that source and structured in the KL Moore style.
Upper Limb — Clinically Oriented Anatomy (Moore-Style)
Based on Gray's Anatomy for Students (the closest available textbook to Moore's COA)
HOW TO READ THIS TUTORIAL
Moore's approach has a consistent pattern for every region:
- Bones — what you're building around
- Joints — how the bones connect and move
- Muscles — what moves what
- Nerves — who controls what
- Blood supply — arteries and veins
- Clinical pearls — what goes wrong and why it matters
We'll follow this pattern through every region of the upper limb, from the shoulder girdle to the fingertips.
REGION 1: THE SHOULDER GIRDLE (Pectoral Girdle)
Think of the shoulder girdle as the foundation platform on which the arm hangs. It connects the arm to the trunk and can be repositioned widely on the chest wall to increase the arm's reach.
Bones
Two bones form the shoulder girdle:
1. Clavicle (Collar Bone)
- The only bony bridge between the arm and the trunk
- S-shaped; medial end is round and thick, lateral end is flat
- You can feel the whole length of it just below your neck
- The medial (sternal) end has a large facet → joins the sternum (SC joint)
- The lateral (acromial) end has a small oval facet → joins the scapula (AC joint)
2. Scapula (Shoulder Blade)
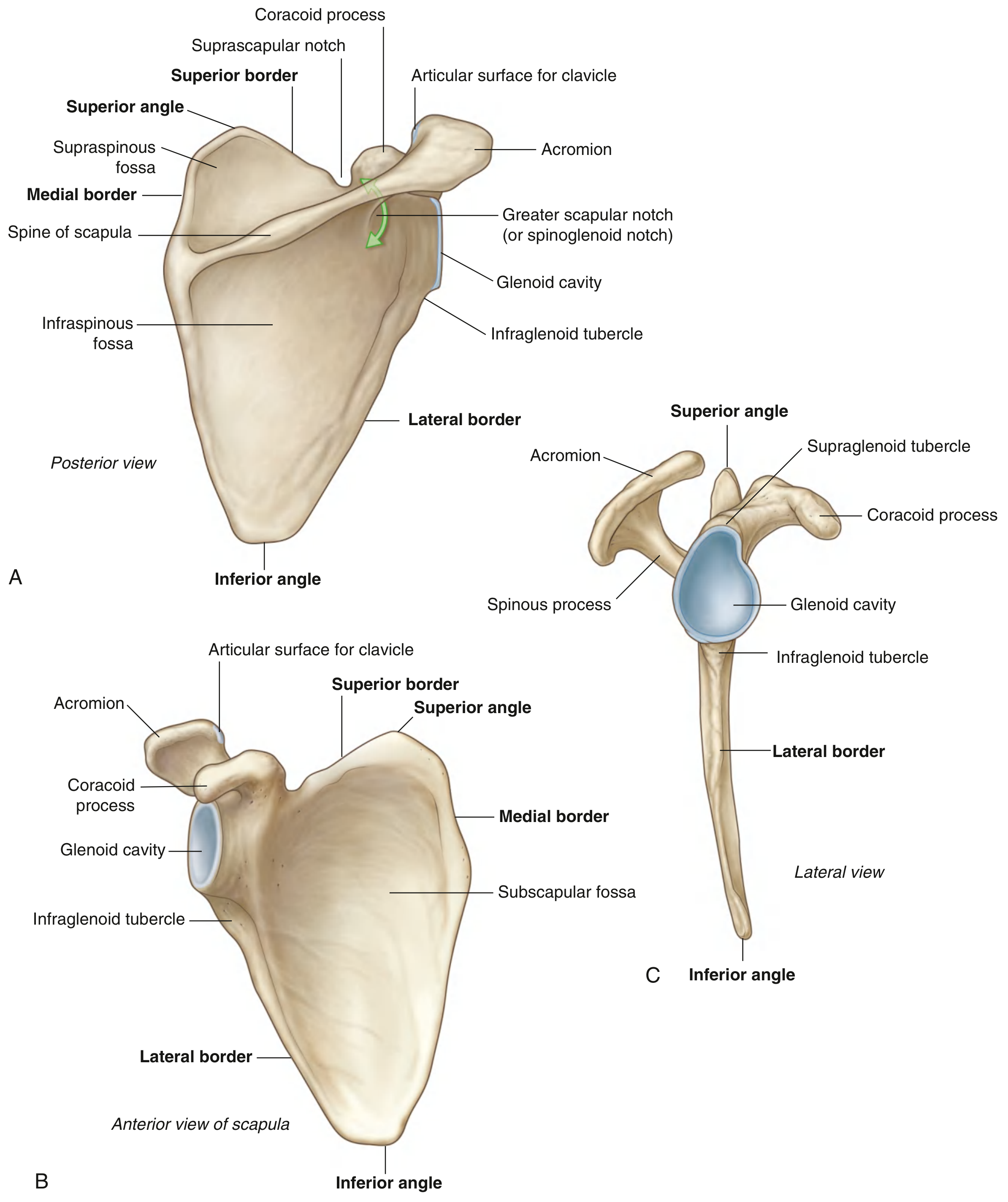
Fig. 7.21 — Right scapula (Gray's Anatomy for Students)
A large, flat, triangular bone sitting on the back of ribs 2–7. Key landmarks:
| Structure | What it is | Why it matters |
|---|---|---|
| Spine | Bony ridge across the back surface | Divides into supraspinous + infraspinous fossae; palpable landmark |
| Acromion | Anterolateral projection of the spine | Arches over glenohumeral joint; articulates with clavicle (AC joint) |
| Coracoid process | Hook-like projection from superior border | Anchor for biceps short head, coracobrachialis, pec minor; palpable under clavicle |
| Glenoid cavity | Shallow cup on lateral angle | Receives the humeral head → glenohumeral joint |
| Supraglenoid tubercle | Small projection above glenoid | Origin of long head of biceps brachii |
| Infraglenoid tubercle | Roughening below glenoid | Origin of long head of triceps brachii |
| Subscapular fossa | Concave anterior (costal) surface | Origin of subscapularis (rotator cuff) |
| Supraspinous fossa | Above the spine (posterior) | Origin of supraspinatus (rotator cuff) |
| Infraspinous fossa | Below the spine (posterior) | Origin of infraspinatus (rotator cuff) |
| Medial border | Thin, sharp inner edge | Origin of serratus anterior |
| Inferior angle | Bottom corner of triangle | Moves forward when arm is raised — palpable landmark |
3. Proximal Humerus
The top end of the upper arm bone. Key landmarks:
| Structure | Description |
|---|---|
| Head | Half-sphere; articulates with glenoid cavity |
| Anatomical neck | Constriction just below the head |
| Greater tubercle | Large projection lateral side → SITS rotator cuff attaches here (except subscapularis) |
| Lesser tubercle | Smaller projection anteriorly → subscapularis attaches here |
| Intertubercular (bicipital) groove | Sulcus between the two tubercles → long head of biceps runs here |
| Surgical neck | Narrowing below the tubercles → most common fracture site (axillary nerve is at risk here) |
Joints of the Shoulder Complex
1. Sternoclavicular (SC) Joint
- Type: Synovial saddle-shaped joint; the only bony connection between the upper limb and the trunk
- Has an articular disc that divides the joint completely into two compartments — acts like a shock absorber
- Reinforced by 4 ligaments: anterior/posterior sternoclavicular, interclavicular, and costoclavicular ligament (the strongest — pins the clavicle to rib 1)
- Movements: clavicle moves in anteroposterior and vertical planes + some rotation
Clinical: SC joint dislocations are rare (only ~3% of shoulder dislocations) but posterior dislocations are dangerous — the medial end of the clavicle can compress the trachea, esophagus, or great vessels.
2. Acromioclavicular (AC) Joint
- Small synovial joint between acromion and lateral clavicle
- Reinforced by: small acromioclavicular ligament (directly over the joint) + large coracoclavicular ligament (conoid + trapezoid parts) — this bigger ligament is the main weight-bearing support
- Allows anteroposterior gliding + axial rotation of scapula
Clinical: AC joint sprain/separation — graded I–III. Grade III: coracoclavicular ligament torn → shoulder drops, clavicle "steps up." Mechanism: fall on tip of shoulder.
Clinical: Clavicle fractures are extremely common (most fractured bone in body). Typically break at the junction of middle and lateral thirds (weakest point). Mechanism: fall on outstretched hand. After fracture: medial fragment pulled UP by sternocleidomastoid; lateral fragment drops DOWN by weight of arm.
3. Glenohumeral (Shoulder) Joint
- Type: Ball-and-socket synovial joint — most mobile joint in the body
- The socket (glenoid cavity) is shallow and only covers about 1/4 of the humeral head → unstable but mobile
- Stability is provided by:
- Glenoid labrum — fibrocartilaginous rim that deepens the socket
- Joint capsule — thin, lax capsule that allows wide movement
- Glenohumeral ligaments — superior, middle, inferior (thickenings of capsule)
- Rotator cuff muscles — the main dynamic stabilizers
- Biceps long head tendon — runs through the joint, helps prevent upward displacement
Movements: flexion, extension, abduction, adduction, medial rotation, lateral rotation, circumduction
Clinical: Shoulder dislocation — most common joint dislocation overall. Usually anterior (95%): humeral head forced forward and inferior. Mechanism: forced external rotation + extension. May injure axillary nerve (test: sensation over deltoid). May cause Bankart lesion (anterior glenoid labrum tear) or Hill-Sachs lesion (posterolateral head compression fracture).
Muscles of the Shoulder
The Rotator Cuff — "SITS"
Four muscles wrap around the glenohumeral joint and are the primary dynamic stabilizers of the shoulder:
| Muscle | Origin | Insertion | Nerve | Action |
|---|---|---|---|---|
| Supraspinatus | Supraspinous fossa | Greater tubercle (top facet) | Suprascapular (C5, C6) | Initiates abduction (first 15°) |
| Infraspinatus | Infraspinous fossa | Greater tubercle (middle facet) | Suprascapular (C5, C6) | Lateral (external) rotation |
| Teres minor | Lateral border of scapula | Greater tubercle (lowest facet) | Axillary (C5, C6) | Lateral rotation |
| Subscapularis | Subscapular fossa | Lesser tubercle | Subscapular nerves (C5, C6, C7) | Medial (internal) rotation |
Clinical: Rotator cuff tears — supraspinatus is the most commonly torn (compressed between acromion and humeral head during abduction). Presents as painful arc from 60–120° of abduction. Subacromial bursa gets pinched in the same space → subacromial impingement.
Other Major Shoulder Muscles
| Muscle | Main Action | Nerve |
|---|---|---|
| Deltoid | Abduction of arm (15–90°); anterior part = flex; posterior = extend | Axillary (C5, C6) |
| Trapezius | Elevates, retracts, and rotates scapula; holds shoulder girdle up | Accessory nerve (CN XI) + C3/C4 |
| Pectoralis major | Adduction, medial rotation, flexion of arm | Medial + lateral pectoral nerves |
| Latissimus dorsi | Powerful extension, adduction, medial rotation ("swimming stroke" muscle) | Thoracodorsal (C6–C8) |
| Serratus anterior | Protracts scapula; holds medial border against ribs; rotates scapula upward | Long thoracic nerve (C5–C7) |
Clinical: Long thoracic nerve injury → paralysis of serratus anterior → winged scapula (medial border lifts off the chest wall when arm is pushed forward against resistance). Caused by: neck surgery, prolonged carrying of heavy bags, viral illness.
REGION 2: THE AXILLA
The axilla (armpit) is the gateway to the upper limb — a pyramidal space where everything passing between the neck/chest and the arm must travel.
Boundaries:
| Wall | Formed by |
|---|---|
| Anterior wall | Pectoralis major (superficial) + pectoralis minor + subclavius (deep) + clavipectoral fascia |
| Posterior wall | Subscapularis (above) + teres major + latissimus dorsi (below) |
| Medial wall | Serratus anterior muscle on ribs 1–4 |
| Lateral wall | Intertubercular groove of humerus (narrowest wall — just a slit) |
| Apex (inlet) | Triangle: clavicle (front) + 1st rib (medial) + scapula superior border (posterior) |
| Floor (base) | Axillary fascia + skin of the armpit |
"Gateways" in the Posterior Wall — Very High Yield
Three spaces between muscles of the posterior wall allow nerves and vessels to exit the axilla:
| Space | Boundaries | What passes through |
|---|---|---|
| Quadrangular space | Teres minor (top) + teres major (bottom) + long head triceps (medial) + surgical neck humerus (lateral) | Axillary nerve + posterior circumflex humeral artery |
| Triangular space | Teres minor (top) + teres major (bottom) + long head triceps (lateral) | Circumflex scapular artery |
| Triangular interval | Teres major (top) + long head triceps (medial) + humerus shaft (lateral) | Radial nerve + profunda brachii artery |
Clinical: Quadrangular space syndrome — compression of axillary nerve here → deltoid and teres minor weakness + "badge area" numbness (lateral arm over deltoid).
Contents of the Axilla
The axilla contains the main neurovascular highway of the upper limb:
- Axillary artery (+ all its branches)
- Axillary vein
- Brachial plexus (cords and terminal branches)
- Axillary lymph nodes (drain breast, arm, thoracic wall — palpated in breast cancer examination)
- Proximal parts of biceps brachii and coracobrachialis muscles
Axillary Artery — 3 Parts (divided by pectoralis minor)
| Part | Branches |
|---|---|
| Part 1 (medial to pec minor) | 1. Superior thoracic artery |
| Part 2 (behind pec minor) | 2. Thoracoacromial artery; 3. Lateral thoracic artery |
| Part 3 (lateral to pec minor) | 4. Subscapular artery (→ circumflex scapular + thoracodorsal); 5. Anterior circumflex humeral; 6. Posterior circumflex humeral |
Mnemonic: "Screw The Lawyer, Save A Patient" (Superior thoracic, Thoracoacromial, Lateral thoracic, Subscapular, Anterior circumflex, Posterior circumflex)
REGION 3: THE BRACHIAL PLEXUS
The brachial plexus is the network of nerves that runs from the neck into the axilla and supplies the entire upper limb (except the skin at the top of the shoulder, which is supplied by C3/C4 supraclavicular nerves).
Formed from: Anterior rami of C5, C6, C7, C8, T1
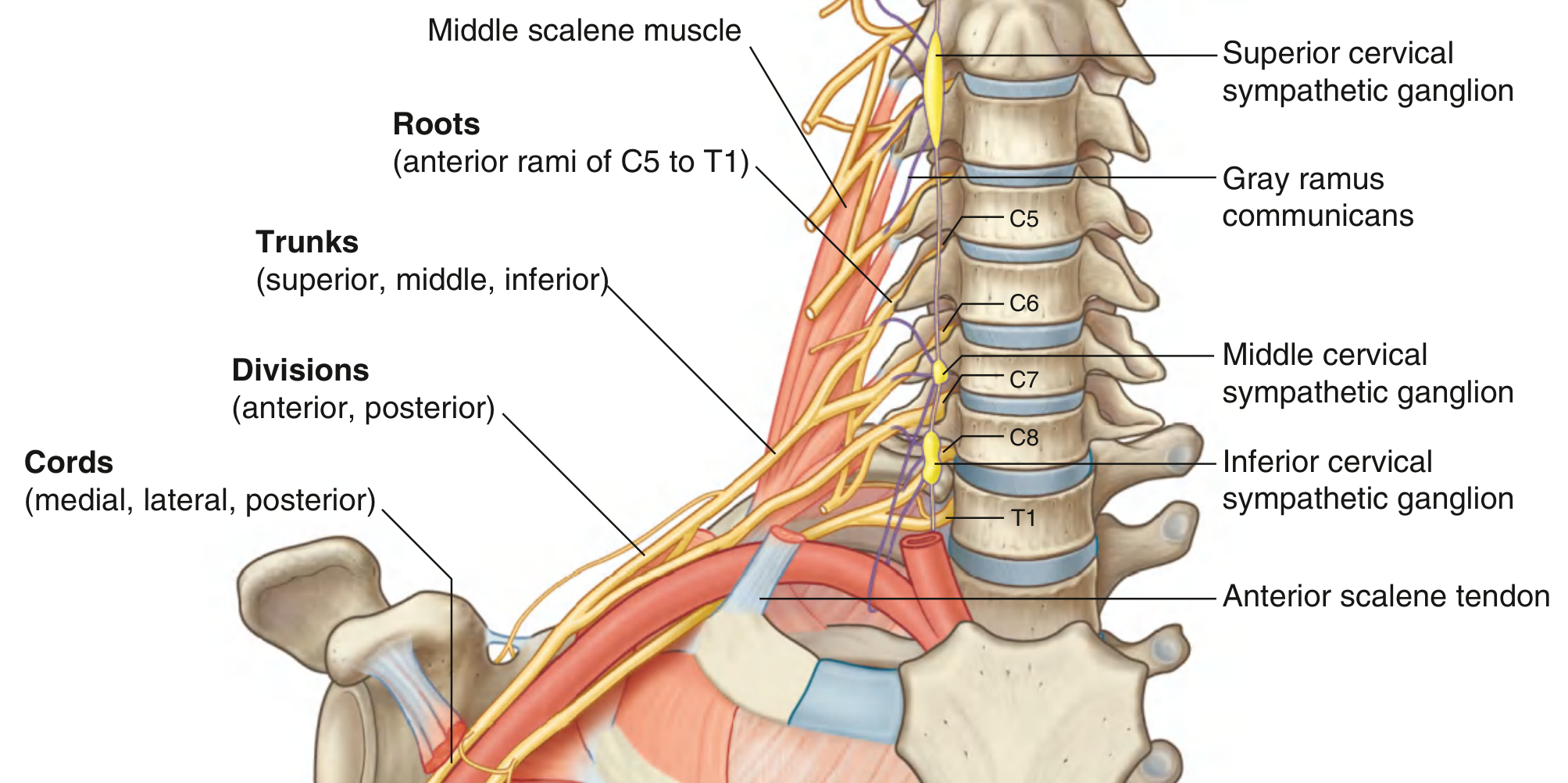
Fig. 7.52 — Brachial plexus from neck into axilla (Gray's Anatomy for Students)
Structure — "Rugby Teams Drink Cold Beer":
| Level | Number | How formed |
|---|---|---|
| Roots | 5 | Anterior rami C5, C6, C7, C8, T1 (pass between anterior and middle scalene muscles) |
| Trunks | 3 | Superior (C5+C6), Middle (C7), Inferior (C8+T1) |
| Divisions | 6 | Each trunk splits into anterior (→ flexors) and posterior (→ extensors) |
| Cords | 3 | Lateral, Medial, Posterior (named by position relative to axillary artery Part 2) |
| Branches | 5 terminal | Musculocutaneous, Median, Ulnar, Radial, Axillary |
Terminal nerves and what they supply:
| Nerve | Root | From | Motor territory | Sensory territory |
|---|---|---|---|---|
| Musculocutaneous | C5, C6 | Lateral cord | Anterior arm (biceps, brachialis, coracobrachialis) | Lateral forearm (as lateral cutaneous n. of forearm) |
| Median | C6–C8, T1 | Lateral + medial cords | Most anterior forearm flexors; thenar muscles; lateral 2 lumbricals | Lateral 3½ digits (palmar), lateral palm |
| Ulnar | C8, T1 | Medial cord | Flexor carpi ulnaris, medial FDP; most intrinsic hand muscles | Medial 1½ digits, medial palm |
| Radial | C5–C8, T1 | Posterior cord | All extensors in arm and forearm; triceps | Posterior arm, posterior forearm, dorsal lateral hand |
| Axillary | C5, C6 | Posterior cord | Deltoid + teres minor | "Badge area" — lateral arm over deltoid |
Clinical Brachial Plexus Injuries:
- Erb's palsy (upper trunk C5–C6): e.g., baby during difficult delivery, motorcycle fall on shoulder. Arm hangs in the "waiter's tip" position — adducted, medially rotated, elbow extended, forearm pronated. Loss of shoulder abduction, elbow flexion.
- Klumpke's palsy (lower trunk C8–T1): e.g., grabbing overhead to prevent a fall. Claw hand (intrinsic muscle paralysis) + Horner's syndrome if T1 rami are involved.
REGION 4: THE ARM (BRACHIUM)
One bone: the humerus shaft. Two compartments.
Compartments of the Arm
| Compartment | Position | Muscles | Nerve | Main action |
|---|---|---|---|---|
| Anterior (flexor) | Front | Biceps brachii, Brachialis, Coracobrachialis | Musculocutaneous | Flex forearm + supinate; flex arm |
| Posterior (extensor) | Back | Triceps brachii (3 heads), Anconeus | Radial | Extend forearm |
Key Muscles in Detail
Biceps brachii:
- Two origins: Long head from supraglenoid tubercle (travels inside the glenohumeral joint capsule, through the bicipital groove); Short head from coracoid process
- Insertion: Radial tuberosity in forearm + bicipital aponeurosis (into deep fascia of forearm)
- Main actions: Powerful supinator of forearm; flexor of forearm at elbow; accessory flexor of arm at shoulder
- Nerve: Musculocutaneous (C5, C6)
- Reflex: Biceps jerk tests C6
Triceps brachii:
- Three heads: Long head (infraglenoid tubercle of scapula), Lateral head (posterior humerus above radial groove), Medial head (posterior humerus below radial groove)
- Insertion: Olecranon process of ulna
- Action: Extend forearm; long head stabilises glenohumeral joint inferiorly
- Nerve: Radial (C7 mainly)
- Reflex: Triceps jerk tests C7
Blood Supply of the Arm
- Brachial artery (continuation of axillary artery, begins at lower border of teres major)
- Gives off profunda brachii (deep brachial artery) → travels with radial nerve in the spiral/radial groove of the humerus → supplies posterior compartment
- Ends at the cubital fossa by dividing into radial and ulnar arteries
Clinical: Fracture of the midshaft of humerus in the spiral groove → radial nerve palsy → "wrist drop" (lost extension of wrist and fingers). Wrist drops; thumb cannot be extended; patient cannot supinate against resistance. Sensation lost over dorsal first web space (only consistent area). Radial nerve palsy is the most common nerve injury from humeral fractures.
REGION 5: THE ELBOW JOINT AND CUBITAL FOSSA
Elbow Joint
The elbow joint is actually three joints sharing one synovial cavity:
| Joint | Bones | Movement |
|---|---|---|
| Humeroulnar joint | Trochlea of humerus + trochlear notch of ulna | Flexion/extension (main hinge) |
| Humeroradial joint | Capitulum of humerus + head of radius | Flexion/extension + forearm rotation |
| Proximal radioulnar joint | Head of radius + radial notch of ulna + anular ligament | Pronation/supination |
Ligaments:
- Ulnar (medial) collateral ligament (UCL) — from medial epicondyle → coronoid process + olecranon; stabilizes against valgus force
- Radial (lateral) collateral ligament (RCL) — from lateral epicondyle → anular ligament
- Anular ligament of radius — holds radial head in the radial notch of the ulna; allows the head to spin during pronation/supination
Clinical — Nursemaid's elbow (pulled elbow): In children < 6 years, sudden pull on the outstretched arm → radial head slips partially out of the anular ligament. The ligament is loose in children. Child holds arm pronated and slightly flexed. Treatment: supination + flexion (the head pops back in).
Clinical — Medial epicondylitis ("Golfer's elbow") vs. Lateral epicondylitis ("Tennis elbow"):
- Tennis elbow: inflammation at common extensor origin at lateral epicondyle; pain on gripping
- Golfer's elbow: inflammation at common flexor origin at medial epicondyle; pain with wrist flexion
Cubital Fossa — The "V" at the Front of Your Elbow
A triangular hollow with:
- Lateral boundary: Brachioradialis muscle
- Medial boundary: Pronator teres muscle
- Roof: Deep fascia + skin (bicipital aponeurosis reinforces the roof)
- Floor: Brachialis + supinator muscles
Contents from lateral to medial — "TAN":
- Tendon of biceps (→ radial tuberosity)
- Artery (brachial artery → divides here into radial and ulnar)
- Nerve (median nerve)
(Radial nerve lies just outside the fossa, lateral under brachioradialis)
Clinical: Brachial artery pulse is felt in the cubital fossa — used for blood pressure measurement (stethoscope bell here when inflating BP cuff).
REGION 6: THE FOREARM (ANTEBRACHIUM)
Two bones: Radius (lateral/thumb side) and Ulna (medial/little finger side)
Connected by:
- Proximal radioulnar joint (elbow level)
- Interosseous membrane (tough fibrous sheet between the bones — transmits forces)
- Distal radioulnar joint (wrist level)
The radius can cross over the ulna → pronation (palm down); uncross → supination (palm up). This is unique to mammals and allows tool use.
Compartments of the Forearm
Anterior Compartment (Flexors) — Innervated mainly by Median nerve (exception: FCU and medial FDP by Ulnar nerve)
Three layers:
| Layer | Muscles | Main action |
|---|---|---|
| Superficial (common origin: medial epicondyle) | Pronator teres, Flexor carpi radialis (FCR), Palmaris longus*, Flexor carpi ulnaris (FCU) | Flex wrist; pronate; FCU = flex + adduct wrist; FCR = flex + abduct wrist |
| Intermediate | Flexor digitorum superficialis (FDS) | Flex middle phalanges of fingers 2–5 (flexes at PIP joint) |
| Deep | Flexor digitorum profundus (FDP), Flexor pollicis longus (FPL), Pronator quadratus | FDP: flex distal phalanges; FPL: flex thumb IP joint; Pronator quadratus: pronation |
*Palmaris longus is absent in ~15% of population. Its tendon is used as a graft in hand surgery.
Clinical — Carpal tunnel syndrome: Median nerve compressed under the flexor retinaculum at the wrist. Presents with numbness/tingling in lateral 3½ fingers (thumb, index, middle, half ring finger), thenar wasting, night pain. Most common nerve compression in the body. Diagnosis: Tinel's sign (tap over carpal tunnel → tingling) and Phalen's test (wrist flexion for 60 sec → symptoms).
Posterior Compartment (Extensors) — Innervated by Radial nerve (deep branch = posterior interosseous nerve)
| Layer | Key muscles | Main action |
|---|---|---|
| Superficial (common origin: lateral epicondyle) | Extensor carpi radialis longus (ECRL), Extensor carpi radialis brevis (ECRB), Extensor digitorum (ED), Extensor carpi ulnaris (ECU), Extensor digiti minimi (EDM) | Extend wrist and fingers |
| Deep | Abductor pollicis longus (APL), Extensor pollicis brevis (EPB), Extensor pollicis longus (EPL), Extensor indicis | Move the thumb and index |
REGION 7: THE WRIST AND HAND
The Wrist (Carpus) — 8 Carpal Bones in 2 Rows
Proximal row (medial to lateral): Pisiform, Triquetrum, Lunate, Scaphoid
Distal row (medial to lateral): Hamate, Capitate, Trapezoid, Trapezium
Mnemonic (proximal to distal, lateral to medial): "Some Lovers Try Positions That They Can't Handle"
= Scaphoid, Lunate, Triquetrum, Pisiform | Trapezium, Trapezoid, Capitate, Hamate
Wrist joint proper = between the distal radius (+ triangular fibrocartilage complex over the distal ulna) and the proximal row of carpals. Movements: flexion, extension, radial deviation (abduction), ulnar deviation (adduction).
Clinical — Scaphoid fracture: Most common carpal fracture. From FOOSH (fall on outstretched hand). Pain in the anatomical snuffbox (see below). Up to 10% have their scaphoid blood supply entering from the distal end only — fracture across the waist cuts off blood to the proximal fragment → avascular necrosis of proximal scaphoid. Danger: X-ray can be normal initially; always treat clinically if snuffbox tenderness is present.
Clinical — Lunate dislocation: FOOSH + hyperextension. The lunate dislocates anteriorly into the carpal tunnel → compresses the median nerve → acute carpal tunnel syndrome.
The Hand
Carpal Tunnel — tunnel formed by the carpal bones (floor/walls) and the flexor retinaculum (roof):
- Contents: 4 tendons of FDS + 4 tendons of FDP + 1 tendon of FPL = 9 tendons, all inside synovial sheaths, + median nerve
- NOT inside: Flexor carpi radialis, ulnar nerve, ulnar artery (these all travel outside the carpal tunnel)
Muscles of the Hand — Intrinsic Muscles:
| Group | Location | Muscles | Main action |
|---|---|---|---|
| Thenar group | Base of thumb (thenar eminence) | Abductor pollicis brevis, Flexor pollicis brevis, Opponens pollicis | Opposition of thumb (touching thumb to fingertips) |
| Hypothenar group | Base of little finger | Abductor digiti minimi, Flexor digiti minimi, Opponens digiti minimi | Move little finger |
| Lumbricals (4) | Palm | Originate from FDP tendons | Flex MCP joints + extend PIP/DIP joints — the "L"-shape movement |
| Interossei (4 dorsal, 3 palmar) | Between metacarpals | Dorsal = abduct fingers; Palmar = adduct fingers | DAB = Dorsal ABducts; PAD = Palmar ADducts |
| Adductor pollicis | Deep palm | Brings thumb toward palm | Adducts thumb (tested by Froment's sign) |
Nerve mnemonic for hand muscles: "LOAF" = Lumbricals 1+2, Opponens pollicis, Abductor pollicis brevis, Flexor pollicis brevis → all supplied by median nerve. Everything else intrinsic → ulnar nerve.
Palmar Arches
The hand has two arterial arches:
| Arch | Formed by | Depth |
|---|---|---|
| Superficial palmar arch | Mainly ulnar artery + small radial contribution | Superficial to flexor tendons; gives common palmar digital arteries → finger supply |
| Deep palmar arch | Mainly radial artery (after entering through 1st dorsal interosseous) + deep ulnar contribution | Deep to tendons; gives palmar metacarpal arteries |
Clinical — Allen's test: Compress both radial and ulnar arteries at wrist, then release one → check hand filling. Tests adequacy of palmar arch anastomosis. Done before radial artery catheterization to ensure the hand won't become ischemic.
Superficial Veins — Clinically Important
- Cephalic vein — originates at lateral (radial) side of dorsal venous network → crosses anatomical snuffbox → runs up lateral forearm and arm → empties into axillary vein in deltopectoral groove
- Basilic vein — originates medial (ulnar) side of dorsal network → runs up medial forearm → pierces deep fascia mid-arm → joins brachial vein → becomes axillary vein
- Median cubital vein — connects cephalic to basilic across the cubital fossa; preferred site for venipuncture (blood draws)
Anatomical Snuffbox
The triangular hollow on the back of the wrist at the base of the thumb when the thumb is extended:
- Medial border: Extensor pollicis longus tendon
- Lateral borders: Extensor pollicis brevis + Abductor pollicis longus tendons
- Floor: Scaphoid + Trapezium bones; distal ends of ECRL and ECRB tendons
- Passes through: Radial artery (deep); Terminal branches of superficial radial nerve (subcutaneous); Origin of cephalic vein
Clinical: Tenderness in the snuffbox after FOOSH = scaphoid fracture until proven otherwise. The radial artery pulse can also be felt here.
MASTER SUMMARY TABLE
| Region | Key bones | Key joints | Key muscles | Key nerves | Key arteries |
|---|---|---|---|---|---|
| Shoulder girdle | Clavicle, Scapula | SC, AC | Trapezius, Serratus anterior, Pec major/minor | Accessory (XI), Long thoracic, Pectoral nerves | Subclavian → Axillary |
| Glenohumeral joint | Proximal humerus | Glenohumeral | Rotator cuff (SITS), Deltoid | Axillary, Suprascapular | Post. circumflex humeral |
| Axilla | — | — | Biceps (short head), Coracobrachialis | Brachial plexus cords + terminal branches | Axillary artery (3 parts, 6 branches) |
| Arm (Brachium) | Humerus shaft | Glenohumeral (above), Elbow (below) | Biceps, Brachialis (ant.); Triceps (post.) | Musculocutaneous (ant.), Radial (post.) | Brachial artery + Profunda brachii |
| Elbow | Distal humerus, Proximal radius, Proximal ulna | Humeroulnar, Humeroradial, Proximal RU joint | Pronator teres, Brachioradialis | Radial, Median, Ulnar all pass here | Brachial → Radial + Ulnar |
| Forearm (Antebrachium) | Radius, Ulna | Radioulnar joints + interosseous membrane | FCR, FCU, FDS, FDP (ant.); Extensors (post.) | Median (ant.), Radial deep branch (post.), Ulnar | Radial artery (lat.), Ulnar artery (med.) |
| Wrist + Hand | 8 carpals, 5 metacarpals, phalanges | Wrist, CMC, MCP, IP | Thenar, Hypothenar, Lumbricals, Interossei | Median (thenar + lateral 3½), Ulnar (rest of hand) | Superficial + deep palmar arches |
CLINICAL SUMMARY — HIGH-YIELD INJURIES BY NERVE
| Nerve injured | Mechanism | Deformity / Sign |
|---|---|---|
| Axillary (C5,C6) | Anterior shoulder dislocation or surgical neck fracture | Cannot abduct arm 15–90°; "badge area" numbness |
| Musculocutaneous (C5,C6) | Coracoid fracture, anterior dislocation | Weak elbow flexion and supination; lateral forearm numbness |
| Radial (C5–C8,T1) | Midshaft humeral fracture (spiral groove) | Wrist drop; cannot extend wrist or fingers; dorsal first web space numb |
| Median (C6–C8,T1) | Supracondylar fracture (distal) or carpal tunnel | "Ape hand" (thenar wasting); cannot oppose thumb; lateral 3½ fingers numb |
| Ulnar (C8,T1) | Medial epicondyle fracture or Guyon's canal compression | "Claw hand" (ring + little finger); cannot abduct/adduct fingers; medial 1½ fingers numb; + Froment's sign |
| Long thoracic (C5–C7) | Neck surgery, carrying heavy loads | Winged scapula (serratus anterior paralysis) |
Source: Gray's Anatomy for Students (ISBN 9780323934237), Chapter 7 — Upper Limb (equivalent to Moore's Clinically Oriented Anatomy regional approach), pp. 821–1066
Create a visual diagram showing brachial plexus anatomy and nerve territories
Generating Image
This is a shared conversation. Sign in to Orris to start your own chat.