H pylori gastritis pathology
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Helicobacter pylori" AND gastritis AND pathology
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive, detailed content from Robbins, Yamada's, and Harrison's, plus histology images and pathway diagrams. I have enough to write a thorough answer.
H. pylori Gastritis - Pathology
The Organism
Helicobacter pylori is a spiral-shaped, gram-negative, microaerophilic bacillus that colonizes the gastric mucous layer, lying close to the gastric foveolar epithelial surface. It was discovered by pathologist Robin Warren and Barry Marshall (Nobel Prize, 2005). The organism has adapted uniquely to the hostile gastric niche through several virulence strategies.
Virulence Factors and Mechanisms of Colonization
H. pylori colonizes via four key structural/enzymatic adaptations (Robbins):
| Factor | Function |
|---|---|
| Flagella | Allow motility in viscous mucus; help the bacterium navigate toward epithelium |
| Urease | Hydrolyzes urea to ammonia, buffering local acidity and protecting the organism from gastric acid |
| Adhesins (BabA, SabA, HopQ) | Bind to Lewis-b antigen and sialyl-Lewis-x on foveolar cells, anchoring the bacterium to the surface epithelium |
| CagA / VacA toxins | Drive mucosal injury and oncogenesis (see below) |
Pathogenesis of H. pylori colonization and mucosal injury (Robbins, Fig. 17.14):
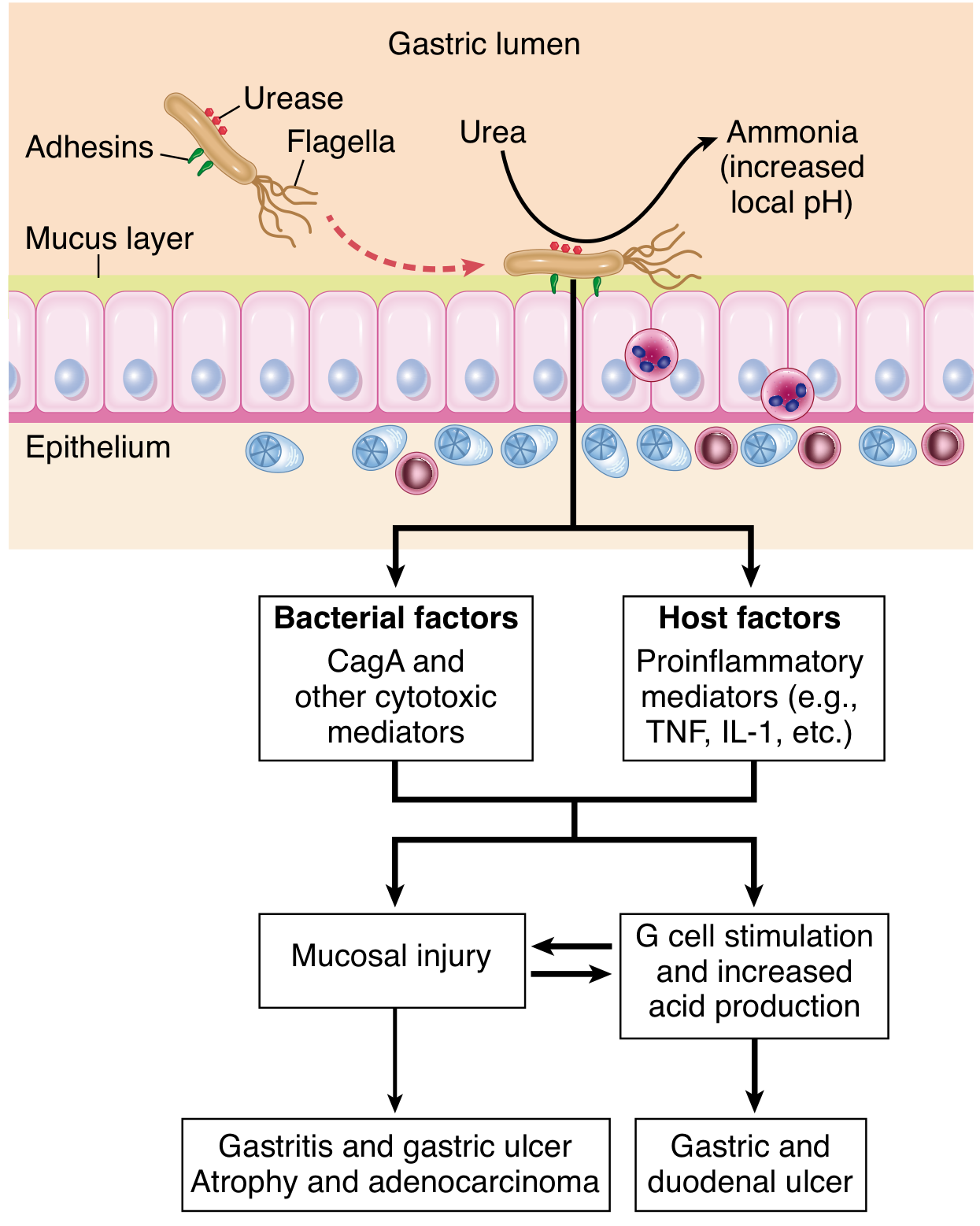
Key Virulence Determinants
cag Pathogenicity Island (cag PAI) and CagA
The cag PAI is present in ~50% of H. pylori isolates globally but in ~90% of isolates in high gastric-cancer-prevalence populations. It encodes a Type IV Secretion System (T4SS) that injects the effector protein CagA directly into gastric epithelial cells. Once inside:
- CagA is tyrosine-phosphorylated by Src/Abl kinases
- Phospho-CagA activates the tyrosine phosphatase SHP-2, causing morphological transformation resembling growth factor stimulation
- Non-phosphorylated CagA interacts with E-cadherin and the c-Met receptor, activating beta-catenin, disrupting cell junctions and polarity
- CagA induces spermine oxidase (SMO), generating oxidative DNA damage and selecting for apoptosis-resistant cells
- CagA also targets p53 in a CagA-dependent manner, modulating apoptosis
The T4SS also translocates peptidoglycan fragments (activating NOD1 -> NF-κB -> IL-8 secretion) and the metabolite heptose bisphosphate (HBP) (activating NF-κB via the TIFA-TRAF6-TAK1 pathway). - Yamada's Textbook of Gastroenterology, 7th ed.
VacA (Vacuolating Cytotoxin A)
- Present in virtually all H. pylori strains but with allelic variation: s1/m1 alleles produce the most vacuolation; s2/m2 produces none
- vacA s1/m1 is strongly associated with peptic ulcer disease and gastric cancer
- VacA induces epithelial vacuolation, apoptosis, and suppresses T-cell responses, contributing to chronic persistence
Outer Membrane Proteins (OMPs)
- BabA binds Lewis-b (Le^b) antigen; babA2+ strains (especially when co-harboring cagA + vacA s1) markedly increase gastric cancer risk
- SabA binds sialyl-Lewis-x, an antigen upregulated during chronic inflammation (bacterial-driven glycosylation remodeling to enhance attachment)
- HopQ binds CEACAM family surface molecules
Immune Response: The Inflammatory Infiltrate
The gastric inflammatory response in H. pylori infection involves (Yamada's):
- Neutrophils - the most distinctive feature; infiltrate the gastric epithelium (intraepithelial neutrophils)
- Lymphocytes and plasma cells (abundant, often in clusters/sheets within the superficial lamina propria)
- Macrophages, eosinophils, and dendritic cells
- Lymphoid follicles with germinal centers (induced MALT)
Cytokine profile: H. pylori drives a dominant Th1 response (IFN-gamma, IL-12). IL-17/Th17 responses are also activated but appear defective in normal hosts, contributing to bacterial persistence. Regulatory T cells (Tregs) expressing FOXP3 are increased in gastric mucosa of infected persons; importantly, infected patients with peptic ulcers have fewer Tregs, while those without ulcers have more, suggesting Treg imbalance shifts clinical outcomes.
H. pylori evades innate immunity by expressing LPS and flagellin that are relatively anergic compared to other enteric bacteria (poor TLR2/4/5 activators). It also expresses arginase, which depletes L-arginine (substrate for iNOS), limiting nitric oxide production.
Histological Morphology (Robbins Pathology, Fig. 17.15)
Gross/endoscopy: Antral mucosa is erythematous with a coarse or nodular appearance; in heavy inflammation, thickened rugal folds may mimic early cancer.
Histology:
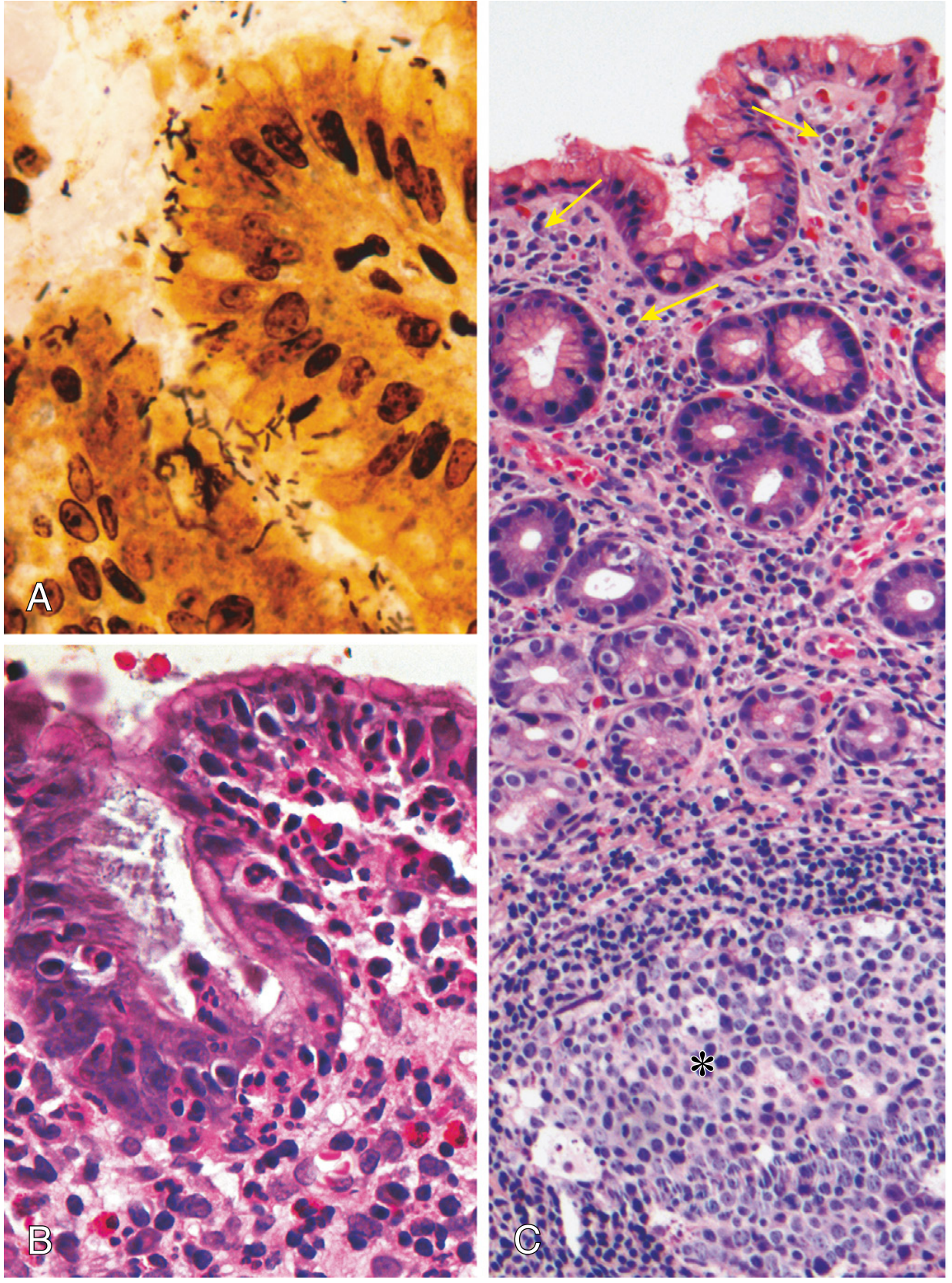
- (A) Spiral/curved organisms visible in the mucus layer; best highlighted by Warthin-Starry silver stain, immunostains, or Giemsa; visible on routine H&E but less obvious
- (B) Dense intraepithelial and lamina propria neutrophils; neutrophils crossing the basement membrane accumulate in gland lumens forming pit abscesses
- (C) Lymphoid aggregates with germinal centers (MALT) in the lamina propria; sheets of plasma cells in the superficial lamina propria - these are hallmarks of H. pylori gastritis
The antrum is the preferred biopsy site (most commonly colonized). In dense infections, organisms spread to the fundus/body.
Patterns of Gastritis and Clinical Outcomes
The topography of inflammation determines disease outcome. This is one of the most clinically important concepts:
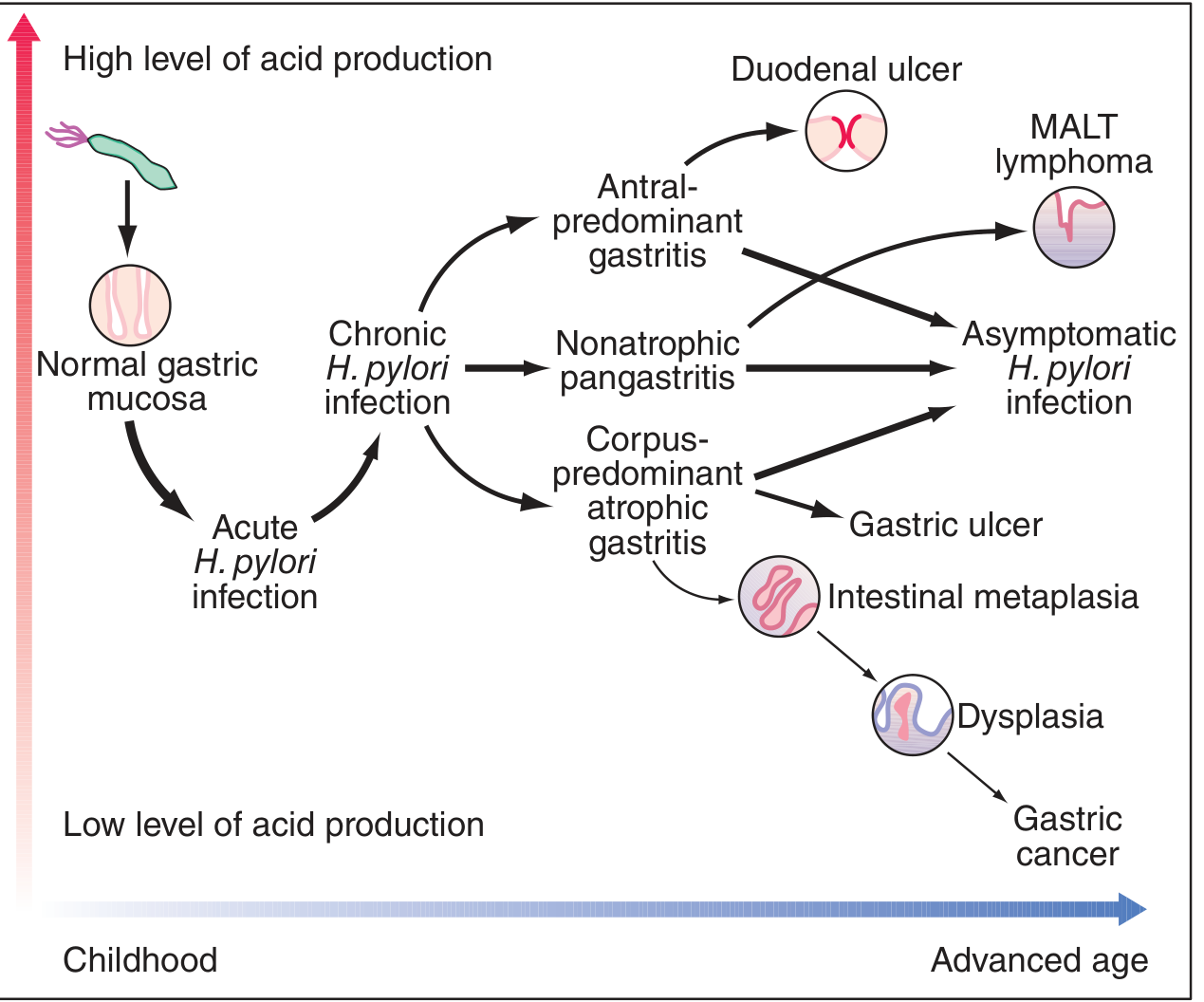
| Gastritis Pattern | Acid Secretion | Outcomes |
|---|---|---|
| Antral-predominant (diffuse antral gastritis) | Normal or increased | Duodenal ulcer, MALT lymphoma |
| Pangastritis (nonatrophic) | Variable | Asymptomatic; can progress |
| Corpus-predominant atrophic gastritis | Decreased | Gastric ulcer, intestinal metaplasia -> dysplasia -> gastric adenocarcinoma |
Key mechanism of increased acid in antral gastritis:
- Antral inflammation suppresses somatostatin (D cells) -> increased gastrin (G cells) -> parietal cell hyperplasia and hypersecretion -> duodenal ulcer risk
- In corpus/pangastritis: inflammation and atrophy destroy parietal cells -> reduced acid -> paradoxically elevated gastrin
Intestinal Metaplasia (IM): Long-standing corpus gastritis leads to replacement of gastric epithelium with intestinal-type epithelium. This is the Correa cascade: chronic gastritis -> atrophy -> IM -> dysplasia -> adenocarcinoma. IM induced by H. pylori is patchy (vs. autoimmune gastritis which is diffuse).
MALT Lymphoma
Lymphoid follicles induced by H. pylori represent acquired MALT. Low-grade gastric MALT lymphoma (MALToma) is driven by antigen-dependent stimulation of B cells by H. pylori. Eradicating H. pylori results in regression of MALT lymphoma in >70% of early-stage cases.
Host Genetic Factors
Polymorphisms affecting disease susceptibility (Robbins, Yamada's):
- TNF and IL-1 promoter polymorphisms -> increased proinflammatory cytokine expression -> pangastritis, atrophy, intestinal metaplasia, cancer
- IL-10 polymorphisms (anti-inflammatory cytokine) -> decreased IL-10 -> same cascade
- Most cag+ infected individuals remain asymptomatic - host genetics are the major determinant of whether peptic ulcer disease or cancer develops
Diagnosis (Biopsy-Based)
| Method | Notes |
|---|---|
| H&E stain | Can see organisms but insensitive |
| Warthin-Starry silver stain / Giemsa | Best for organism visualization on biopsy |
| Rapid urease test (CLO test) | Detects urease activity in biopsy specimen; fast and cheap |
| Immunostain | Highly sensitive for H. pylori antigen |
| Culture / PCR | Used for antibiotic resistance testing |
| Urea breath test | Non-invasive; detects ^13C/^14C-labeled CO2 after urease splits labeled urea |
| Fecal antigen test | Non-invasive; good for confirmation of eradication |
Summary: Sequence of H. pylori Mucosal Injury
- Colonization - urease buffers pH; flagella navigate mucus; adhesins anchor the bacterium
- Acute injury - ammonia and toxins (CagA, VacA) directly damage epithelium
- Innate immune activation - NF-κB activation via NOD1 (peptidoglycan), TIFA (HBP), TLR9 (DNA) -> IL-8 -> neutrophil recruitment
- Adaptive immune activation - Th1 skewing (IFN-gamma dominant); Treg and Th17 modulation
- Chronic gastritis - plasma cells, lymphoid follicles, MALT formation
- Atrophy and intestinal metaplasia - in corpus-predominant disease
- Neoplasia - gastric adenocarcinoma (Correa cascade) or MALT lymphoma
Sources:
- Robbins & Cotran Pathologic Basis of Disease, 10th ed. - Figs 17.14, 17.15
- Yamada's Textbook of Gastroenterology, 7th ed. - Sections on H. pylori virulence and gastritis patterns
- Harrison's Principles of Internal Medicine, 22nd ed. (2025) - Fig. 335-8
Recent evidence note: A 2026 APAGE practice guideline (PMID: 41057234) recommends surveillance for H. pylori-associated gastric premalignant conditions (atrophy, IM, dysplasia), reinforcing the importance of the Correa cascade outlined above.
This is a shared conversation. Sign in to Orris to start your own chat.