why is sucrose test a screening test for PNH
Sucrose Test as a Screening Test for PNH
The Principle
- CD55 (Decay Accelerating Factor) inactivates C3/C5 convertase complexes
- CD59 (Membrane Inhibitor of Reactive Lysis) blocks assembly of the membrane attack complex (C5b-9)
Why It Is a Screening Test (Not Confirmatory)
| Feature | Sucrose Test | Ham's Test (Acidified Serum) |
|---|---|---|
| Sensitivity | High (sensitive) | High (sensitive) |
| Specificity | Lower (false positives) | Higher |
| Role | Screening | Confirmatory |
| Mechanism | Low ionic strength activates complement mildly | Acidified serum activates complement via alternative pathway |
| False positives | Megaloblastic anemia, autoimmune hemolytic anemia | Fewer |
An Important Distinguishing Point
- PNH → sucrose test positive
- HEMPAS → sucrose test negative — Quick Compendium of Clinical Pathology, 5th ed.
Current Status
how sucrose activates compliment ?
How Sucrose "Activates" Complement in the Sucrose Test
1. Low Ionic Strength from Sucrose Solution
2. Effect of Low Ionic Strength on Complement
- The electrostatic repulsion between complement proteins and the cell surface is reduced
- This allows complement proteins to bind more readily to cell membranes
- Specifically, it promotes C3 binding and deposition onto the RBC surface via the alternative pathway's "tick-over" mechanism — the slow, spontaneous hydrolysis of C3 into C3(H₂O) that happens continuously in plasma
3. Why Normal Cells Are Unaffected
- CD55 degrades C3 convertase (C3bBb) — stopping the amplification loop
- CD59 blocks the membrane attack complex (MAC, C5b-9) from inserting into the membrane
4. Why PNH Cells Lyse
- The modest C3b deposition triggered by low ionic strength goes unchecked
- The alternative pathway amplification loop runs freely
- MAC (C5b-9) assembles on the RBC membrane
- The cell lyses → hemolysis
Analogy
Key Point for Exams
g6pd pathophyzuology
G6PD Deficiency — Pathophysiology
The Normal Role of G6PD
Glucose-6-phosphate + NADP⁺ → 6-phosphogluconolactone + NADPH + H⁺
H₂O₂ + 2 G-SH → 2 H₂O + G-S-S-G
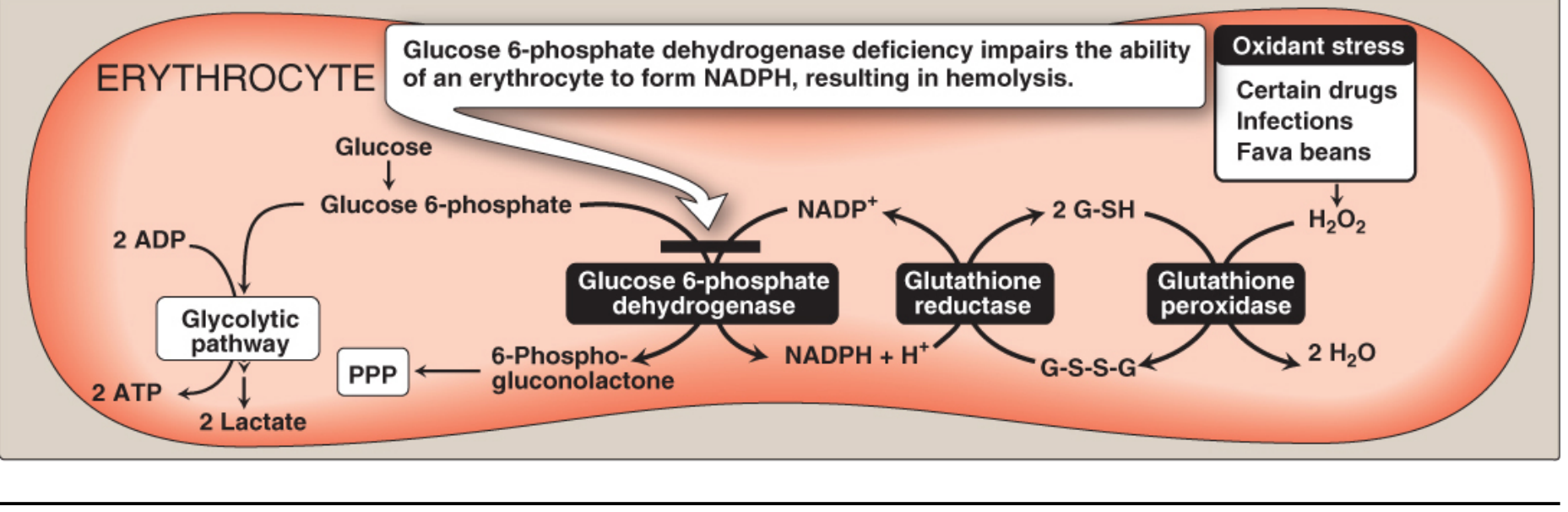
Why RBCs Are Uniquely Vulnerable
- The PPP is the ONLY source of NADPH in RBCs. Other cells have alternative NADPH-generating pathways (e.g., malic enzyme). RBCs do not.
- RBCs have no nucleus or ribosomes — they cannot synthesize new G6PD enzyme. As the existing enzyme degrades (especially unstable mutant variants), protection against oxidative stress is irreversibly lost.
What Happens in G6PD Deficiency
| Step | Event |
|---|---|
| 1 | Oxidant stress → H₂O₂ and free radicals generated inside RBC |
| 2 | G6PD is deficient → NADPH cannot be regenerated |
| 3 | Without NADPH → G-SH pool is depleted |
| 4 | H₂O₂ accumulates → oxidizes hemoglobin sulfhydryl groups |
| 5 | Oxidized Hb denatures → forms Heinz bodies (insoluble precipitates attached to RBC membrane) |
| 6 | Membrane proteins are also oxidized → membrane rigidity increases |
| 7 | Rigid, Heinz body-laden RBCs are trapped and destroyed by macrophages in the spleen → extravascular hemolysis |
| 8 | Severe cases → intravascular hemolysis as well |
Precipitating Factors
- Oxidant drugs — sulfonamides, dapsone, primaquine, nitrofurantoin, rasburicase
- Fava beans (favism) — contain vicine and convicine, which are oxidant compounds; particularly associated with the Mediterranean G6PD variant
- Infections — macrophages generate free radicals during the inflammatory response; these diffuse into RBCs causing oxidative damage
G6PD Variants and Severity
| Class | Residual Enzyme Activity | Clinical Features |
|---|---|---|
| I | <10% | Chronic non-spherocytic hemolytic anemia (CNSHA) |
| II* | <10% | Severe acute hemolytic anemia (e.g., Mediterranean variant) |
| III* | 10–60% | Moderate (e.g., G6PD A−, common in Africa) |
| IV | >60% | Asymptomatic |
Inheritance
Evolutionary Note
The enzyme deficiency test is usually not done during an active state, but it is done after resolution of an active phase, or it may be performed weeks after the hemolytic episode results. That's because during an active state, all the G6PD deficiency enzymes are hemestep and it doesn't show properly or what is the reason, I forgot.
Why G6PD Testing Is Unreliable During an Acute Hemolytic Episode
The Core Reason: Selective Destruction of the Most Deficient Cells
- The oldest, most G6PD-deficient RBCs are selectively destroyed first (they have the least enzyme activity and cannot withstand the oxidant load)
- The bone marrow responds by releasing a surge of reticulocytes and young RBCs into the circulation
- Young RBCs naturally have higher G6PD activity — even in G6PD-deficient individuals, young cells haven't yet run down their enzyme supply
The Result: A False-Normal Test
"During an acute hemolytic episode, the assay would measure the enzyme activity present in reticulocytes where there are usually normal levels, resulting in a falsely normal assay." — Creasy & Resnik's Maternal-Fetal Medicine
"The oldest, most G6PD-deficient red cells have been selectively destroyed, and young red cells, having higher G6PD activity, are being released into the circulation. Under these conditions, only a quantitative test can give a definitive result." — Harrison's Principles of Internal Medicine, 22nd ed.
When to Test
- Wait 2–3 months after the hemolytic episode, when the RBC population has normalized and older cells (with low G6PD) are again present in adequate numbers
- If you must test urgently, use a quantitative assay (not a screening/spot test), which is more sensitive and may still detect deficiency even in a reticulocyte-rich sample
- DNA-based testing is not affected by the RBC age distribution and can diagnose at any time
Summary in One Sentence
to write in mz note one line above concept '
Why parvovirus B19 infects progenitor erythroid cells or red cells and causes a temporary halt of erythropoiesis, usually leading to significant anemia in a setting of a pre-existing marrow stress, maybe like sickle cell anemia or something like that? Can you explain it a bit, why it's unaffected only progenitor red cells and why not progenitor white blood cells or platelet cells?
Why Parvovirus B19 Selectively Targets Erythroid Progenitors
The Receptor is the Key: P Antigen (Globoside)
- Erythroid progenitors and erythroblasts — highest expression → primary target
- Megakaryocytes — express globoside too (more on this below)
- Endothelial cells
- Fetal myocardial cells
- Myeloid progenitors (neutrophil/monocyte lineage)
- Lymphoid progenitors
Why Does It Halt Erythropoiesis?
- The cell ceases to proliferate and is lysed
- Erythroid output from the marrow drops to near zero
- Reticulocytes disappear from the peripheral blood within days
- This is called pure red cell aplasia (PRCA) — only the red cell lineage is affected
Why It Becomes Catastrophic in Underlying Hemolytic Conditions
- RBCs are being destroyed much faster than normal (lifespan reduced from 120 days to sometimes 10–20 days)
- The bone marrow is already running at maximum compensatory overdrive — producing red cells 6–8× the normal rate just to maintain baseline hemoglobin
- There is no reserve capacity
The hemoglobin crashes rapidly and severely → Transient Aplastic Crisis (TAC)
What About Platelets and White Cells?
| Lineage | P Antigen Expression | Effect of B19 |
|---|---|---|
| Erythroid progenitors | High | Directly lysed → aplasia |
| Megakaryocytes (platelet precursors) | Present | Can be infected, but less efficiently |
| Myeloid progenitors (WBC) | Absent/minimal | Not infected |
| Lymphoid progenitors | Absent | Not infected |
One-Line Summary for Notes
MONOSPOT OR heterophile agglutation test explainn why heterophile ? Inf mononucleosis
Monospot / Heterophile Agglutination Test in Infectious Mononucleosis
What Does "Heterophile" Mean?
Why Does EBV Cause Heterophile Antibodies?
- Human EBV-infected cells
- Red blood cells of sheep, horses, and cattle
Key point: heterophile antibodies are not directed against EBV — they are a byproduct of nonspecific B cell activation. They do not correlate with disease severity and are not protective. — Sherris & Ryan's Medical Microbiology, 8th ed.
How the Monospot Test Works
- A drop of patient serum is placed on a card/slide
- It is first mixed with guinea pig kidney cells — these absorb away cross-reacting antibodies from serum sickness and other conditions (specificity step)
- Then horse RBCs (or sheep RBCs in older versions) are added
- If heterophile antibodies are present → they agglutinate the horse RBCs → positive result
Why Called "Heterophile Agglutination"?
| Term | Meaning |
|---|---|
| Heterophile | Antibodies that react with antigen from a different species |
| Agglutination | Clumping/aggregation of RBCs by the antibody |
Performance of the Test
| Feature | Value |
|---|---|
| Sensitivity | ~75–85% (adults); only ~40% in children; <20% in children <4 yrs |
| Specificity | ~90% |
| Antibody type | IgM |
| Appears | Week 1–2 of illness |
| Peaks | Week 2–3 |
| Persists | Up to 3–6 months |
One-Line Summary for Notes
Cd34= means hematopoietic stem cells, Cd55 meand DAF CD19 and IL5 , CD21 iam so confudssed can you create a notes with all this imp markers amd what i shou;ld know to siolve usmle question immunolgy
USMLE CD Marker Master Notes
HOW TO THINK ABOUT CD MARKERS
T CELL MARKERS
| Marker | Cell | What to Know |
|---|---|---|
| CD3 | All T cells | Forms complex with TCR. Pan-T cell marker — if it's a T cell, it has CD3. Loss of CD3 = no T cell signaling |
| CD4 | Helper T cells | Binds MHC II. Also the receptor HIV gp120 binds (with CCR5/CXCR4 as co-receptor). CD4 count monitors HIV progression |
| CD8 | Cytotoxic T cells | Binds MHC I. Kills virus-infected and tumor cells |
| CD28 | All T cells (CD4 > CD8) | Costimulatory receptor on T cells. Binds B7 (CD80/86) on APCs → Signal 2 for T cell activation. Without it → T cell anergy |
| CD40L (CD154) | Activated CD4⁺ T cells | Binds CD40 on B cells → triggers B cell class switching. Hyper-IgM syndrome = CD40L deficiency → can't switch from IgM to IgG/IgA/IgE |
| CD25 | Activated T cells & Tregs | IL-2 receptor α-chain. Tregs = CD4⁺CD25⁺FoxP3⁺ |
| CD45RA | Naïve T cells | "RA = Raw/new" |
| CD45RO | Memory T cells | "RO = Old/memory" |
| CD1 | Developing T cells, Langerhans cells | Presents lipid antigens (like mycobacterial antigens) — MHC I-like |
| CD2 | T cells, NK cells | Adhesion molecule; marker of T cell activation |
| CD7 | T cells, stem cells | Marker for T-cell ALL and stem cell leukemia |
B CELL MARKERS
| Marker | Cell | What to Know |
|---|---|---|
| CD19 | All B cells (not plasma cells) | Pan-B cell marker. Part of B cell co-receptor complex (with CD21 + CD81). Target of rituximab in lymphoma treatment |
| CD20 | Pre-B → mature B cells (not plasma cells) | Rituximab target. Lost when B cell becomes plasma cell (that's why rituximab doesn't kill plasma cells) |
| CD21 | Mature B cells, follicular dendritic cells | CR2 = Complement receptor 2 — binds C3d. Also the EBV receptor (EBV binds CD21 to enter B cells → infectious mononucleosis) |
| CD40 | B cells, macrophages, dendritic cells | Receives signal from CD40L on T helper cells → class switching |
| CD10 | Pre-B cells, germinal center B cells | Marker for ALL (B cell) and follicular lymphoma |
| CD5 | T cells + some B cells (B-1 cells) | B cells expressing CD5 → seen in CLL and mantle cell lymphoma |
| CD23 | B cells, macrophages, eosinophils | Low-affinity IgE receptor (FcεRII). Elevated in CLL (CLL = CD5⁺ CD23⁺) |
| CD38 | Early B and T cells, plasma cells | Marker for plasma cells and multiple myeloma. Target of daratumumab |
| CD138 (Syndecan-1) | Plasma cells | Definitive plasma cell marker. Positive in multiple myeloma |
NK CELL MARKERS
| Marker | Cell | What to Know |
|---|---|---|
| CD16 | NK cells, macrophages | FcγRIII — binds IgG Fc → mediates ADCC (antibody-dependent cellular cytotoxicity) |
| CD56 | NK cells | Pan-NK marker. Also seen in NK/T cell lymphoma |
| CD57 | NK cells, some T cells | Mature NK cells |
MYELOID / MONOCYTE MARKERS
| Marker | Cell | What to Know |
|---|---|---|
| CD14 | Monocytes, macrophages | LPS (endotoxin) co-receptor. GPI-anchored — absent in PNH |
| CD15 | Granulocytes | Marker for Reed-Sternberg cells in Hodgkin lymphoma (CD15⁺ CD30⁺) |
| CD30 | Activated T/B cells | Reed-Sternberg cells in Hodgkin lymphoma (CD15⁺ CD30⁺). Target of brentuximab vedotin |
| CD64 | Monocytes, macrophages | High-affinity FcγRI |
| CD11b / CD18 | Neutrophils, macrophages | Integrin — mediates adhesion. Deficiency → LAD (Leukocyte Adhesion Deficiency) |
STEM CELL / PROGENITOR MARKERS
| Marker | Cell | What to Know |
|---|---|---|
| CD34 | Hematopoietic stem cells | The HSC marker. Used to identify and isolate stem cells for transplant. Stains HSC-derived tumors (e.g., some AML, GIST also CD34⁺) |
| TdT (Terminal deoxynucleotidyl transferase) | Pre-T, Pre-B cells | Enzyme in immature lymphocytes. Positive in ALL, negative in mature lymphomas |
COMPLEMENT REGULATORY PROTEINS (GPI-anchored — critical for PNH)
| Marker | Cell | What to Know |
|---|---|---|
| CD55 (DAF — Decay Accelerating Factor) | All blood cells | Degrades C3/C5 convertase → stops complement amplification. Absent in PNH → complement-mediated hemolysis |
| CD59 (MIRL — Membrane Inhibitor of Reactive Lysis) | All blood cells | Blocks MAC (C5b-9) assembly. Absent in PNH — most important protein protecting RBCs. GPI-anchored |
| CD35 (CR1) | RBCs, B cells, monocytes | Complement receptor 1 — binds C3b/C4b → phagocytosis |
PNH high-yield: PNH = loss of all GPI-anchored proteins (due to PIG-A mutation) → CD55 and CD59 absent → uncontrolled MAC → intravascular hemolysis. Flow cytometry showing absent CD55/CD59 = gold standard diagnosis.
ADHESION / TRAFFICKING MARKERS
| Marker | Cell | What to Know |
|---|---|---|
| CD11a/CD18 (LFA-1) | Leukocytes | Binds ICAM-1. Required for leukocyte extravasation. Absent in LAD type I |
| CD31 (PECAM-1) | Endothelial cells, platelets | Mediates leukocyte migration through vessel walls |
| CD62L (L-selectin) | Naïve lymphocytes | Homing receptor for lymph nodes — naïve T cells use this to enter lymph nodes |
CYTOKINE RECEPTORS (USMLE-RELEVANT)
| Marker | Cytokine | Key Disease |
|---|---|---|
| CD25 | IL-2 receptor α | Tregs = CD4⁺ CD25⁺ FoxP3⁺ |
| CD122 | IL-2/IL-15 receptor β | NK and memory T cell survival |
| CD127 | IL-7 receptor | Critical for T cell development in thymus |
| CD132 | Common γ chain (γc) | Shared by IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 receptors. X-linked SCID = γc deficiency → no T, B (dysfunctional), NK cells |
IL-5 receptor: on eosinophils and basophils. IL-5 = the eosinophil growth/activation cytokine. IL-5 blockade (mepolizumab) treats severe eosinophilic asthma.
DISEASE-MARKER QUICK LOOKUP
| Disease / Scenario | Key Markers |
|---|---|
| T-ALL | TdT⁺, CD7⁺, CD3⁺, CD4⁺CD8⁺ (double positive) |
| B-ALL | TdT⁺, CD19⁺, CD10⁺ (CALLA), CD20⁺ |
| CLL | CD5⁺, CD19⁺, CD20(dim)⁺, CD23⁺, TdT⁻ |
| Mantle Cell Lymphoma | CD5⁺, CD19⁺, CD20⁺, CD23⁻, Cyclin D1⁺ |
| Follicular Lymphoma | CD10⁺, CD19⁺, CD20⁺, BCL-2⁺ |
| Multiple Myeloma | CD138⁺, CD38⁺, CD19⁻, CD20⁻ |
| Hodgkin Lymphoma (RS cells) | CD15⁺, CD30⁺, CD45⁻ |
| PNH | CD55⁻, CD59⁻ on RBCs/WBCs |
| LAD | CD11b/CD18⁻ (integrin absent) |
| X-linked SCID | CD132 (γc) deficiency |
| Hyper-IgM syndrome | CD40L (CD154) deficiency on T cells |
| HIV target cell | CD4⁺ T cell (gp120 binds CD4 + CCR5/CXCR4) |
| EBV entry into B cells | CD21 (EBV receptor) |
| Rituximab target | CD20 (B cells in lymphoma/RA) |
ONE-LINE MNEMONICS
- CD3 = TCR's partner (always together, always T cell)
- CD4 = "4 letters in HELP" → helper; also HIV's door
- CD8 = "8 = kill" → cytotoxic killer
- CD19/20 = B cell's ID card (lost when becoming plasma cell)
- CD21 = EBV + C3d receptor (2 jobs, 1 marker)
- CD34 = Stem cell marker (think: "34 = stem")
- CD55/59 = PNH's missing bodyguards
- CD40L = T cell's signal to B cell ("switch now!") → absent = Hyper-IgM
we have generally have a high leukocyte concentration, which will lead to generalized erythema against acute compared to chronic erythrocytosis, we have a filamented there, with the bulb is enlarged rather than white bulb in contrast to all other chronic leukemias. And, yeah, can you break that down Hy sticky notes usmle
🗒️ STICKY NOTES: Acute vs Chronic Leukemia — USMLE Smear Findings
CONCEPT 1 — Why Acute Leukemia Has HIGH WBC + Leukostasis
- Blasts are large, rigid, sticky → clog microcirculation
- Symptoms: hypoxia, dyspnea (pulmonary), confusion/seizures (CNS), visual changes
- AML gets leukostasis at lower counts than ALL (myeloblasts are bigger/stickier than lymphoblasts)
- CLL patients can tolerate WBC >500,000 without leukostasis — mature small lymphocytes are small and deformable
USMLE tip: Young patient, very high WBC, confusion + hypoxia = leukostasis → emergency leukapheresis
CONCEPT 2 — The Blast Cell: What That "Large Nucleus With Prominent Nucleolus" Means
| Feature | Blast (Acute) | Mature cell (Chronic) |
|---|---|---|
| Nucleus size | Large — fills most of the cell | Smaller, condensed |
| Nuclear chromatin | Fine/open (euchromatin — active transcription) | Coarse/clumped (heterochromatin) |
| Nucleolus | Prominent, visible (1–3 nucleoli) | Absent or inconspicuous |
| N:C ratio | High — nucleus dominates, scant cytoplasm | Low — more cytoplasm |
| Maturity | Immature — stuck, cannot differentiate | Mature — functionally useless but morphologically complete |
The "bulb" you described = the prominent nucleolus inside the large open nucleus of the blast. This is the hallmark of active, rapidly dividing, undifferentiated cells. The nucleolus = ribosome factory = cell is in overdrive growth mode.
CONCEPT 3 — Auer Rods (AML ONLY — never in ALL)
- Pink/red needle-like rods in cytoplasm of myeloblasts
- Made of fused primary azurophilic granules (myeloperoxidase-positive)
- Pathognomonic for AML — if you see them, it's AML, period
- Most abundant in APL (AML M3, t(15;17)) — bundles of Auer rods = "faggot cells"
- APL + Auer rods = DIC risk (granules release procoagulants)
USMLE: Blast with rod-shaped pink cytoplasmic inclusion = AML → check for DIC
CONCEPT 4 — Smudge Cells (CLL — Chronic)
- CLL lymphocytes are fragile → smear preparation crushes them → smudge/basket cells
- Seen in CLL (and sometimes Waldenström's)
- The opposite of blasts — these cells are mature but mechanically fragile
- CLL smear = small mature lymphocytes + smudge cells
USMLE: Elderly patient, WBC 80,000, small lymphocytes, smudge cells on smear = CLL
CONCEPT 5 — The Core Difference: Acute vs Chronic at a Glance
| Acute Leukemia | Chronic Leukemia | |
|---|---|---|
| Cell type | Blasts (immature, >20% in marrow) | Mature but clonal cells |
| Smear | Large cells, open chromatin, nucleoli, high N:C | Small/mature cells, condensed chromatin |
| WBC | Very high, very sick | Can be extremely high but asymptomatic longer |
| Onset | Sudden, weeks | Insidious, months-years |
| Leukostasis risk | HIGH (especially AML) | Low (CLL tolerates 500K+) |
| Special finding | Auer rods (AML) | Smudge cells (CLL) |
| Treatment urgency | Emergency | Often "watch and wait" |
ONE-LINE EACH FOR NOTES
- Blast = large nucleus + prominent nucleolus + scant cytoplasm + fine chromatin = immature = ACUTE leukemia
- Auer rod = pink needle in cytoplasm = AML only → rule out APL (DIC risk)
- Smudge cell = crushed fragile lymphocyte on smear = CLL
- Leukostasis = WBC >100K + blasts (AML worse than ALL) → plugs microvessels → CNS + lung emergency
- Chronic leukemia cells = mature-looking, condensed chromatin, no nucleolus, no Auer rods
Here I asked my presenter if you should give me a bit of chronic leukemia and rather chronic leukemia with hairy leukemia in general in PLL or in chronic leukemia, there will be an increased accumulation of the white blood cells, which is the lymphocytes in the white pulp of spleen and splenomegaly also present because all the cells get trapped with the lymphatic macrophages. And specifically, but here is the leukemia, there is no lymphadenopathy, and accumulation of hairy cells in the red pulp, why
🗒️ STICKY NOTES: Hairy Cell Leukemia vs CLL — Spleen, Lymph Nodes & Why Red Pulp
FIRST — The Normal Spleen Architecture (to understand the difference)
| Zone | What lives there normally |
|---|---|
| White pulp | Lymphocytes (T and B cells) — the immune zone |
| Red pulp | RBCs, macrophages, sinusoids — the filtration/blood zone |
| Marginal zone | Transition between the two |
WHY HAIRY CELLS GO TO THE RED PULP (not white pulp)
Red pulp is massively infiltrated → white pulp is obliterated → spleen looks grossly beefy red (not the white-on-red pattern of CLL)
WHY NO LYMPHADENOPATHY IN HAIRY CELL LEUKEMIA
- Normal lymph node trafficking requires CCR7 (responds to CCL19/CCL21 chemokines secreted in lymph nodes)
- Hairy cells do not express CCR7 → they cannot respond to the "come to the lymph node" signal
- Without CCR7, they cannot enter lymph nodes via high endothelial venules (HEVs)
- So lymph nodes are spared
USMLE high-yield: Massive splenomegaly + NO lymphadenopathy = think Hairy Cell Leukemia first
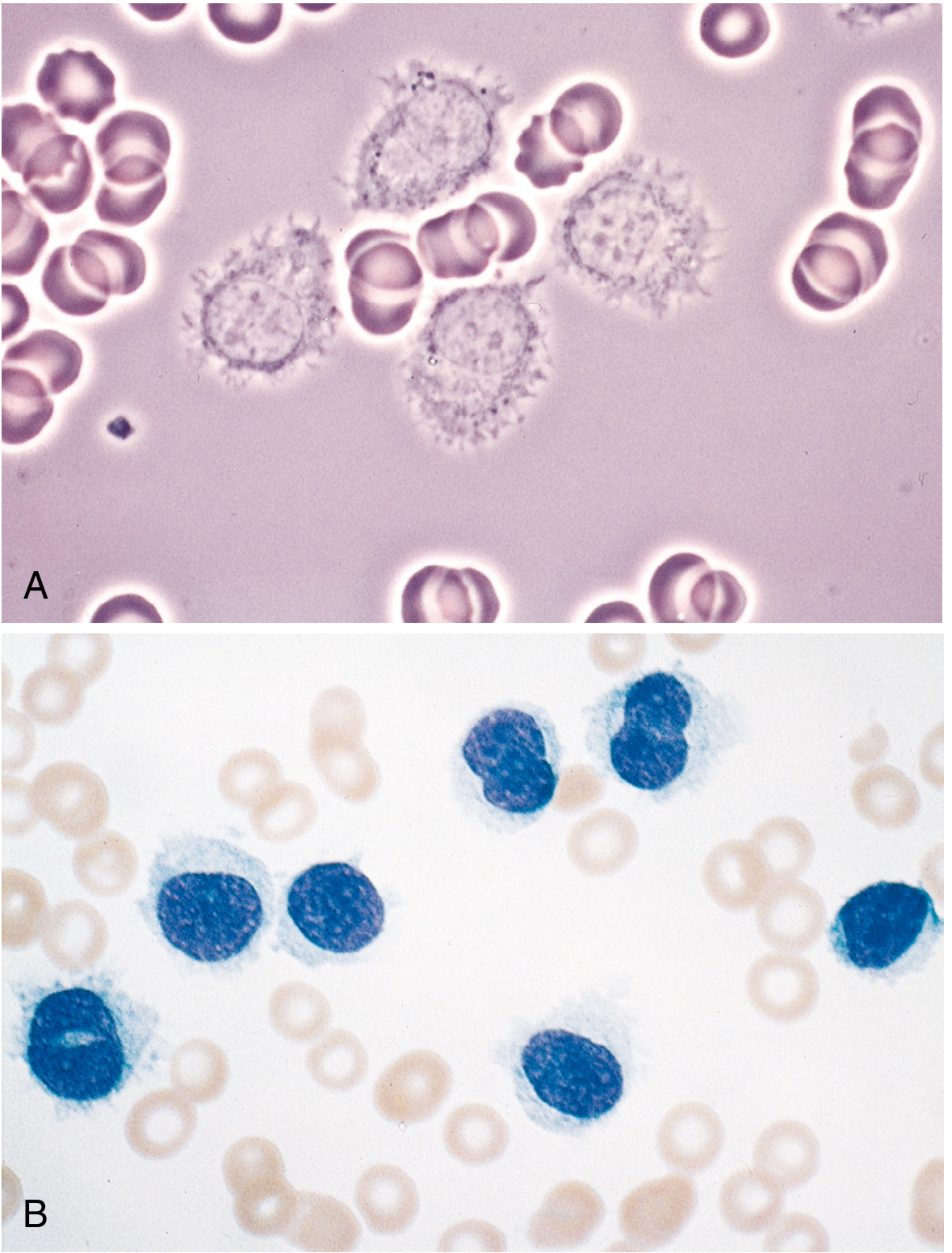
CLL vs HCL vs PLL COMPARISON TABLE
| Feature | CLL | Hairy Cell Leukemia (HCL) | PLL (Prolymphocytic Leukemia) |
|---|---|---|---|
| Cell origin | Mature B cell | Mature B cell | Mature B cell (or T) |
| WBC count | Very HIGH (lymphocytosis) | LOW / pancytopenia | Very HIGH |
| Spleen | Enlarged (white pulp) | Massively enlarged (RED pulp) | Enlarged |
| Lymphadenopathy | Yes — prominent | No — absent/rare | Variable |
| Smear finding | Small round lymphocytes + smudge cells | Cells with hairy cytoplasmic projections, oval/reniform nucleus | Large cells with prominent single central nucleolus (>55% of cells) |
| Bone marrow | Infiltrated, aspiratable | Infiltrated + fibrosis → "dry tap" | Infiltrated |
| Monocytes | Normal | Monocytopenia (unique!) | Normal |
| Special stain | — | TRAP positive (tartrate-resistant acid phosphatase) | — |
| CD markers | CD5⁺, CD19⁺, CD20(dim)⁺, CD23⁺ | CD19⁺, CD20⁺, CD11c⁺, CD25⁺, CD103⁺, CD5⁻, CD23⁻ | CD19⁺, CD20(bright)⁺, FMC7⁺ |
| Mutation | del(13q), del(11q), +12 | BRAF V600E (>90%) | — |
| Treatment | Ibrutinib, venetoclax | Cladribine (purine analogue), BRAF inhibitor (vemurafenib) | — |
WHY PANCYTOPENIA IN HCL (not just lymphocytosis like CLL)
- Bone marrow infiltration → less space for normal hematopoiesis
- Reticulin fibrosis (hairy cells secrete fibronectin) → marrow becomes fibrotic → dry tap → can't produce normal cells
- Hypersplenism → enlarged spleen traps and destroys RBCs, platelets, neutrophils
Monocytopenia specifically = unique to HCL among all leukemias. The mechanism isn't fully understood but is very characteristic. It explains why HCL patients get atypical mycobacterial infections (monocytes/macrophages are needed to contain mycobacteria).
🗒️ ONE-LINE STICKY NOTES
- HCL = Red pulp spleen (hairy cells home to sinusoids via CD11c/integrins, not white pulp follicles)
- No lymphadenopathy in HCL = no CCR7 → can't enter lymph nodes
- Dry tap = hairy cells + reticulin fibrosis → bone marrow can't be aspirated
- TRAP stain positive = HCL (tartrate-resistant acid phosphatase)
- BRAF V600E = HCL mutation (same as melanoma)
- Monocytopenia = unique to HCL → atypical mycobacteria risk
- CD103 positive = HCL marker (along with CD11c, CD25)
- CLL spleen = white pulp; HCL spleen = red pulp — always on USMLE