Can you explain pediatric tumor from robbins
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from Robbins & Kumar Basic Pathology. Let me compile the full detailed answer.
Pediatric Tumors - Robbins & Kumar Basic Pathology
(Robbins & Kumar Basic Pathology, Chapter 4 - Pediatric Diseases)
Overview
Malignant neoplasms are the second most common cause of death in children between ages 4 and 14 years - only accidents exact a higher toll. Benign tumors are even more common than malignancies.
Tumorlike Lesions (Not True Neoplasms)
Before discussing true tumors, two special categories must be recognized:
1. Heterotopia (Choristoma)
- Microscopically normal cells or tissues present in abnormal locations
- Examples: ectopic pancreatic tissue in the stomach wall, adrenal cells in the kidney, lungs, or ovaries
- Usually of little clinical significance but can be confused with neoplasms
2. Hamartoma
- Excessive but focal overgrowth of cells and tissues native to the organ in which it occurs
- Mature cellular elements are identical to the normal organ but lack normal architecture
- The line between hamartoma and benign neoplasm is often unclear - both can be clonal
- Examples: hemangiomas, lymphangiomas, rhabdomyomas of the heart, adenomas of the liver
- Though histologically benign, can cause serious complications due to size or location
Benign Neoplasms
Three benign tumors deserve special mention:
1. Hemangioma
- The most common neoplasm of infancy
- Both cavernous and capillary types occur; the latter are often more cellular than those in adults and may appear deceptively worrisome
- Predominantly in the skin - particularly face and scalp - as irregular, erythematous to violaceous lesions ranging from flat to plaque to nodular forms
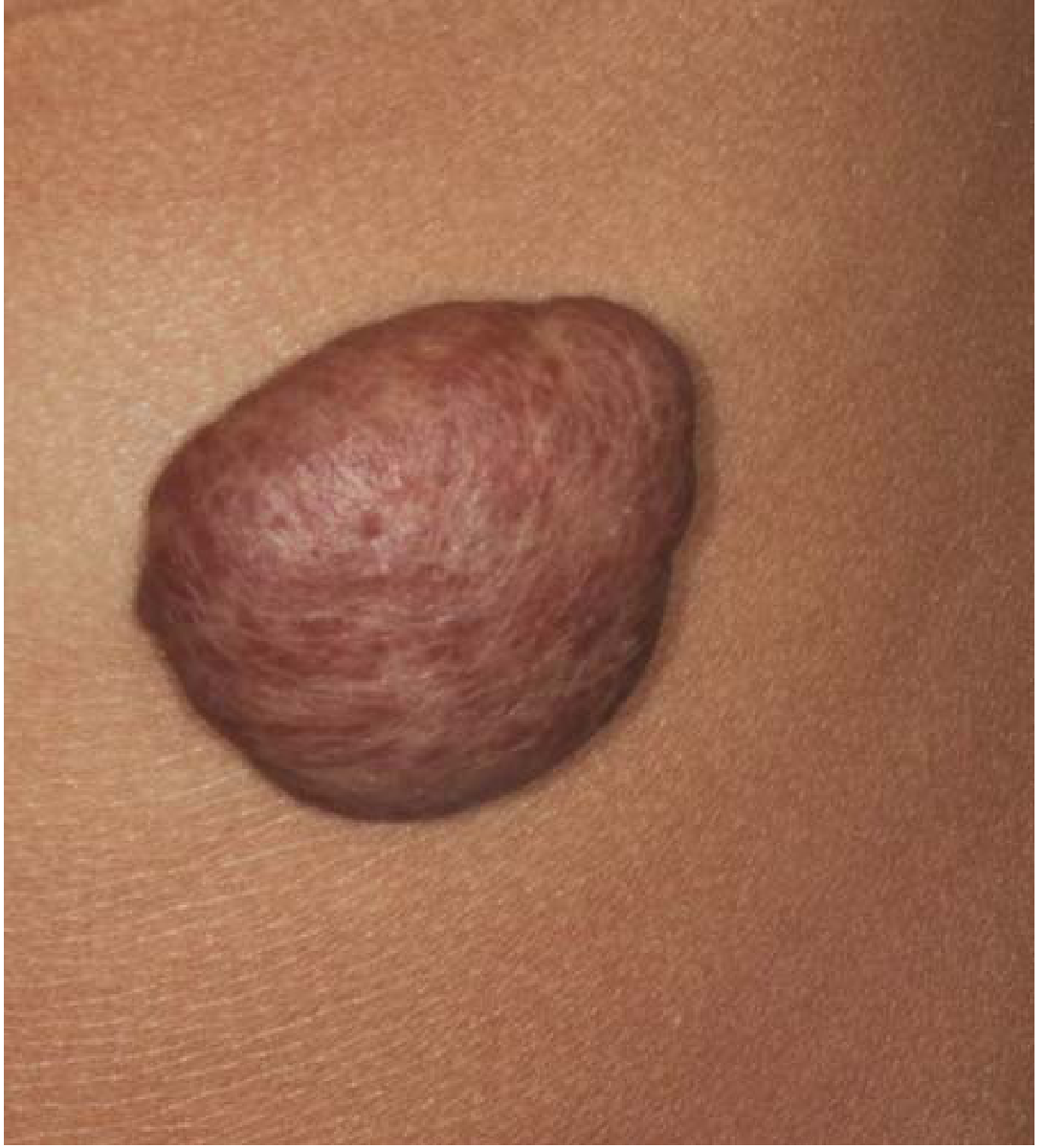
- Hemangiomas may enlarge during the first few years of life but most spontaneously regress by age 5-8 years
- If needed, treatment includes laser therapy, beta-blockers, or surgery
2. Lymphangioma
- Benign proliferations of lymphatic channels
- Can occur superficially in the skin or in deeper locations (mouth, neck, axilla, mediastinum, retroperitoneum)
- Deep lesions may cause serious clinical complications due to their location
3. Teratoma
- Arise from totipotent cells and contain elements of all three germ layers
- Most common site: sacrococcygeal region (Fig. 4.37 below); also gonads, retroperitoneum, mediastinum
- The most common tumors of infancy in the first few months of life
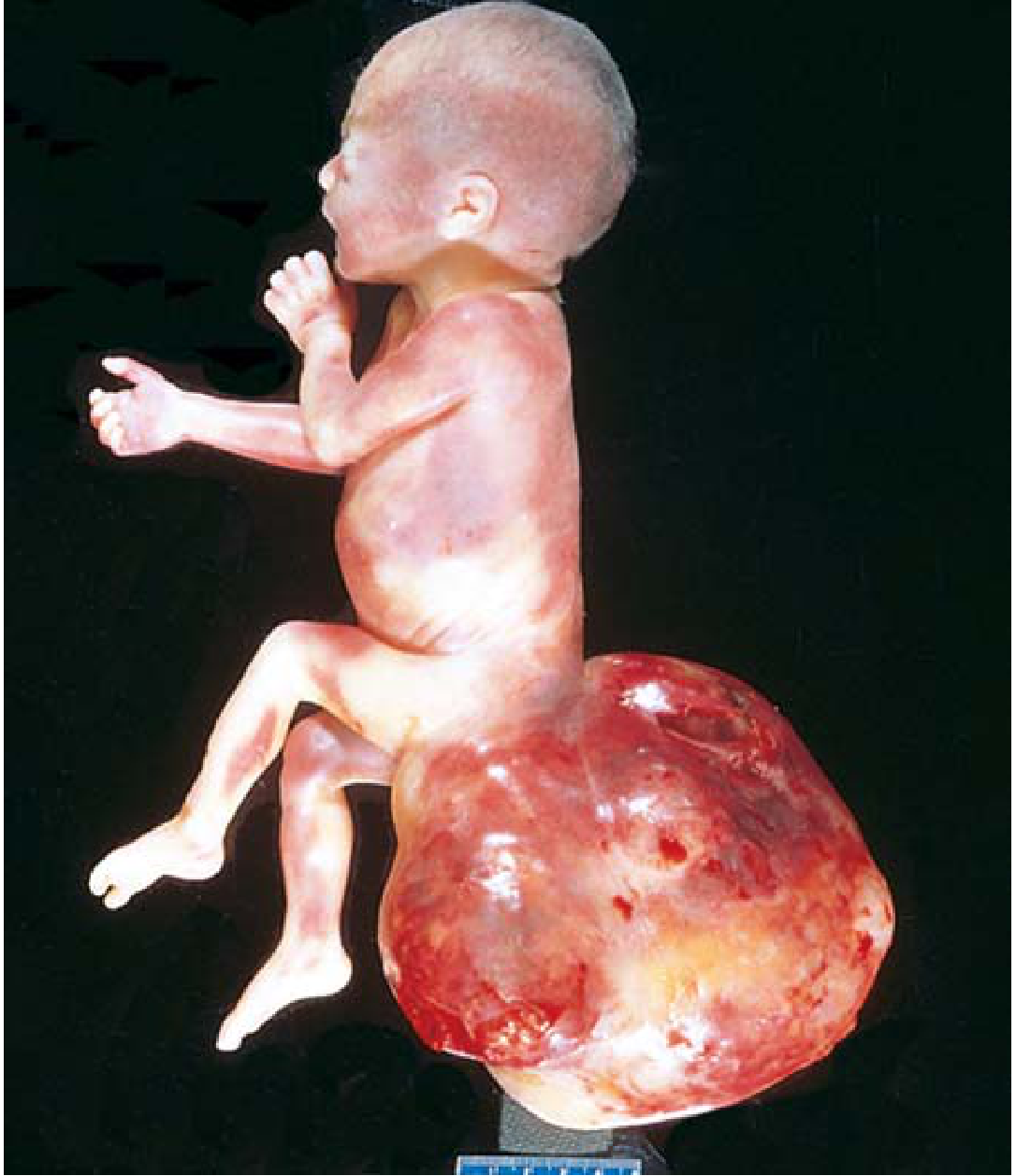
- Sacrococcygeal teratomas: ~75% are mature (benign); malignant teratomas are more common in male infants
- Malignant potential depends on age: those presenting in infancy tend to be benign; those presenting later are more likely malignant
- Associated with elevated alpha-fetoprotein (AFP) if malignant elements are present
Malignant Neoplasms
Key Differences from Adult Cancers
| Feature | Pediatric Cancers | Adult Cancers |
|---|---|---|
| Most common systems involved | Hematopoietic, nervous system, soft tissues | Lung, breast, prostate, colon (epithelial) |
| Genetic basis | Frequent germline mutations | Predominantly somatic mutations |
| Relationship to development | Close link between teratogenesis and oncogenesis | Less common |
| Spontaneous regression | Can occur (especially neonatal) | Extremely rare |
| Histology | Primitive/embryonal (small, round, blue cells) | Pleomorphic-anaplastic |
| Prognosis | Many are curable | Often less favorable |
"Small, Round, Blue Cell Tumors" of Childhood
A distinctive category - sheets of cells with small, round nuclei. Includes:
- Neuroblastoma
- Lymphoma
- Rhabdomyosarcoma
- Ewing sarcoma
- Medulloblastoma
- Retinoblastoma
- Some cases of Wilms tumor
Confirmatory molecular studies are routinely used for diagnosis and prognosis.
The Three Classic Pediatric Malignancies
1. Neuroblastoma
The most common extracranial solid tumor of childhood. Arises from neural crest cells of the adrenal medulla or sympathetic ganglia.
Epidemiology:
- Median age at diagnosis: ~2 years; 90% diagnosed before age 5
- Most cases are sporadic; ~1-2% are familial (ALK mutations, germline)
Sites:
- Adrenal medulla (~40%)
- Paraspinal sympathetic ganglia (abdomen, posterior mediastinum, pelvis, neck)
Genetics:
- MYCN amplification - present in ~25% of cases; strongly associated with poor prognosis
- 1p deletion (loss of tumor suppressor locus)
- 17q gain
- ALK mutations (familial cases)
Morphology:
- Sheets of small, round, blue cells with dark nuclei and scant cytoplasm
- Homer-Wright pseudorosettes: tumor cells arranged around a central neuropil
- Variable degrees of neuroblastic differentiation
Clinical Features:
- Abdominal mass (most common presentation) that may cross the midline
- Hypertension (from catecholamine secretion)
- Elevated urinary catecholamines: VMA (vanillylmandelic acid) and HVA (homovanillic acid) - used for diagnosis and monitoring
- "Blueberry muffin" appearance - subcutaneous metastatic nodules in neonates
- Periorbital ecchymosis ("raccoon eyes") from orbital metastases
- Paraneoplastic syndrome: opsoclonus-myoclonus ("dancing eyes, dancing feet")
- Stage 4S disease (special stage in infants <1 year): liver, skin, and bone marrow involvement but WITHOUT bone metastases - can spontaneously regress
Prognosis:
- Age and stage are the strongest prognostic factors
- Infants (<18 months): favorable prognosis even with metastatic disease (especially 4S)
- Older children: poor prognosis with metastatic disease
- MYCN amplification = poor prognosis regardless of stage
- Trk-A (high neurotrophin receptor expression) = favorable prognosis
- 5-year survival: ~50% overall; >90% in low-risk, ~20% in high-risk
2. Wilms Tumor (Nephroblastoma)
The most common primary renal tumor of children. A prototype of the link between oncogenesis and aberrant embryogenesis.
Epidemiology:
- Peak age: 3-4 years
- Bilateral in ~5-10% of cases
- Associated syndromes:
- WAGR syndrome - Wilms tumor, Aniridia, Genitourinary anomalies, intellectual disability (mental Retardation); deletion of WT1 gene (chromosome 11p13)
- Denys-Drash syndrome - Wilms tumor, gonadal dysgenesis, nephropathy; point mutation in WT1
- Beckwith-Wiedemann syndrome - organomegaly, macroglossia, hemihypertrophy, Wilms tumor; associated with WT2 locus (11p15.5) or IGF2 overexpression
- WTX gene mutations (on X chromosome) - present in ~30% of sporadic cases
Precursor Lesion: Nephrogenic rests - microscopic clusters of primitive metanephric blastema cells in the renal parenchyma; present in ~40% of Wilms tumors
Morphology:
- Classic triphasic pattern:
- Blastemal component - sheets of small, undifferentiated cells (worst prognosis)
- Stromal component - spindle-shaped cells
- Epithelial component - primitive tubules/glomeruli
- Gross: large, solitary, encapsulated mass that distorts the kidney; gray-tan cut surface with areas of hemorrhage, cyst, necrosis
- Anaplasia (nuclear enlargement with multipolar mitotic figures) is the most important adverse histologic feature
Clinical Features:
- Large abdominal/flank mass, usually unilateral and detected incidentally by parents or doctor
- Hematuria, hypertension, abdominal pain
- The mass rarely crosses the midline (contrast with neuroblastoma)
Genetics:
- WT1 tumor suppressor gene (11p13) - involved in genitourinary development
- WT2 (11p15.5) - associated with Beckwith-Wiedemann syndrome
- WTX gene mutation (~30% sporadic cases)
- Mutations in CTNNB1 (β-catenin gene) in ~15% of cases
Prognosis:
- Excellent overall - one of the great success stories of oncology
- 5-year survival: >90% with combined surgery + chemotherapy +/- radiotherapy
- Anaplastic histology confers worse prognosis
- Stage remains the most important prognostic factor
3. Retinoblastoma
- Most common intraocular tumor of children
- Arises from immature retinal cells (retinoblasts)
- The classic example of the "two-hit hypothesis" of tumor suppressor gene inactivation (RB1 gene, chromosome 13q14)
- Hereditary form: one germline mutation + one somatic mutation; often bilateral and multifocal; autosomal dominant inheritance pattern
- Sporadic form: two somatic mutations; usually unilateral, unifocal
- Clinical: leukocoria ("cat's eye reflex" - white pupillary reflex), strabismus, visual loss
- Morphology: small, round blue cells with Flexner-Wintersteiner rosettes and fleurettes
- Spontaneous regression via calcification can occur (leading to "retinocytoma")
- Treatment: chemotherapy, laser photocoagulation, cryotherapy, radiation, enucleation in advanced cases
- Germline RB1 carriers have markedly increased risk of secondary malignancies (osteosarcoma, soft tissue sarcomas) - "second-hit" in other tissues
Summary Table
| Tumor | Age | Key Gene | Key Feature | Prognosis |
|---|---|---|---|---|
| Hemangioma | Infancy | - | Most common benign tumor; spontaneous regression | Excellent |
| Teratoma | Infancy/fetal | - | Sacrococcygeal; germ cell origin | Mostly benign; malignant in older males |
| Neuroblastoma | <5 years | MYCN | Adrenal/sympathetic; VMA/HVA elevated; Homer-Wright pseudorosettes | Variable - excellent in infants, poor with MYCN amplification |
| Wilms Tumor | 3-4 years | WT1, WT2, WTX | Renal; triphasic; WAGR/Denys-Drash/BWS associations | >90% 5-year survival |
| Retinoblastoma | <5 years | RB1 | Two-hit hypothesis; leukocoria; bilateral = hereditary | Generally good; risk of 2nd malignancies in hereditary form |
Source: Robbins & Kumar Basic Pathology, Chapter 4 - Pediatric Diseases, "Tumors and Tumorlike Lesions of Infancy and Childhood" (pp. 143-149)
This is a shared conversation. Sign in to Orris to start your own chat.