Solitary fibrous tumour of bone
Reading File
Finding Sources
Finding Sources
Searching PubMed
"solitary fibrous tumor" AND bone
Finding Sources
Finding Sources
Searching PubMed
"solitary fibrous tumor" AND bone AND STAT6
Reading File
Reading File
Reading File
Reading Article
Reading Article
Reading File
Finding Sources
Finding Sources
Searching PubMed
"solitary fibrous tumor" AND "primary bone" OR "intraosseous solitary fibrous"
Reading Article
Reading Article
Reading Article
Reading File
Loading Image
I now have comprehensive data to deliver a thorough, well-structured answer on solitary fibrous tumour of bone.
Solitary Fibrous Tumour of Bone
Overview
Solitary fibrous tumour (SFT) is a rare fibroblastic mesenchymal neoplasm originally described in the pleura and now recognized across virtually all anatomical sites. Primary intraosseous (bone) SFT is extremely rare - only a few dozen well-documented cases exist in the literature. It was historically often misclassified as hemangiopericytoma, fibrosarcoma, or even Ewing sarcoma. The 2020 WHO Classification of Soft Tissue and Bone Tumours recognizes intraosseous SFT as a distinct entity with intermediate-to-malignant biological potential.
Terminology and Classification
- The term hemangiopericytoma (HPC) has been unified with SFT since both share the defining NAB2::STAT6 gene fusion on chromosome 12q13.
- Under the 2020 WHO classification, extrameningeal SFT (including bone) is listed as intermediate (rarely metastasizing) or malignant.
- For bone specifically, the WHO 2020 update recognizes intraosseous SFT as a primary bone tumour category.
Molecular Biology - The Defining Alteration
The hallmark is a cryptic inversion of chromosome 12q13 fusing the NAB2 and STAT6 genes:
- NAB2::STAT6 fusion is found in ~83-100% of cases (RT-PCR or NGS)
- The most common fusion variants are NAB2ex4-STAT6ex2 and NAB2ex6-STAT6ex16/17
- The fusion converts NAB2 (a transcriptional repressor of EGR1) into a transcriptional activator, driving oncogenesis via EGR1 target genes
- The chimeric STAT6 protein translocates to the cell nucleus, which is detectable by IHC - this is the basis of the diagnostic STAT6 nuclear staining
Additionally, TP53 variants were found in 91.6% of primary bone SFT cases in one institutional series (Bianchi et al., 2021 - PMID 34069378), a rate markedly higher than in soft tissue SFTs, potentially explaining the more aggressive behaviour seen in bone primaries.
Epidemiology
| Feature | Detail |
|---|---|
| Incidence | Extremely rare; only few dozen bone cases in world literature |
| Age | Typically 4th-6th decade (mean ~53 years); wide range |
| Sex | No clear predilection |
| Common sites (bone) | Flat and short bones predominate; skull, mandible, pelvis, vertebrae, proximal femur (intertrochanteric) |
Clinical Presentation
- Often asymptomatic until large or until pathological fracture occurs
- May present with pain, swelling, or an incidental radiological finding
- Pathological fracture can be the presenting event (as in the case of proximal femur intertrochanteric SFT)
- Paraneoplastic hypoglycaemia (Doege-Potter syndrome) - due to IGF-2 secretion - is rare but reported even with bone primaries
- Bone SFTs tend to be slowly growing but can behave aggressively independent of conventional histological parameters
Imaging
Plain Radiograph / CT
- Lytic, well-demarcated intramedullary lesion
- May show cortical thinning, expansion, or destruction
- Calcifications occasionally present
- No specific periosteal reaction is typical
- Differential on imaging is broad: Ewing sarcoma, fibrosarcoma, GCT, metastasis, plasmacytoma
MRI
- Heterogeneous signal - fibrous areas hypointense on T2, vascular areas enhance avidly
- Rich vascularisation produces prominent contrast enhancement
- Helps define soft tissue extension
A key point: imaging is not diagnostic and bone SFTs are frequently misdiagnosed radiologically. Initial biopsy readings of Ewing sarcoma have been reported due to this non-specificity.
Gross Pathology
SFTs are typically well-circumscribed, firm, whitish-grey masses with a whorled cut surface and dense fibrous texture. Viscid cysts may be present. The gross specimen below (from a pleural SFT) illustrates the characteristic whorled fibrous appearance seen across sites:
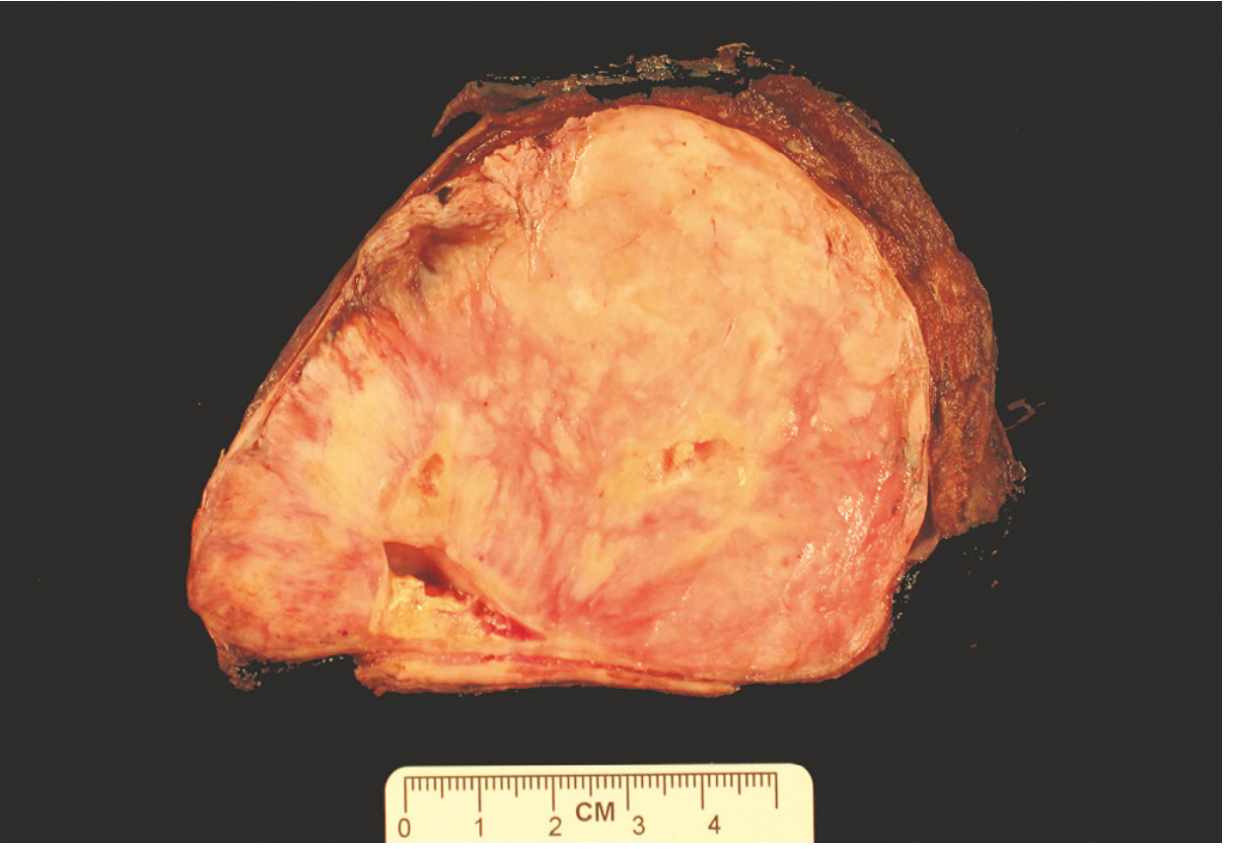
Gross cut surface of a SFT showing solid, whorled fibrous appearance (Robbins Pathologic Basis of Disease)
Histopathology
Classic Features
- "Patternless pattern" - haphazardly arranged spindle-to-oval cells with indistinct cell borders
- Dense collagen bands admixed with the tumour cells
- Characteristic branching, staghorn (haemangiopericytoma-like) thin-walled vessels - ectatic, ramifying
- Finely dispersed chromatin; cells have plump or tapered nuclei with lightly eosinophilic cytoplasm
- Alternating hypercellular and hypocellular areas
Malignant Features (any 2 of the following raise concern)
- High cellularity with cytologic atypia
- Mitotic activity >4 mitoses/10 HPF
- Necrosis
- Invasive growth pattern
Important caveat for bone SFTs: The Bianchi et al. 2021 institutional series found that 15/20 bone SFT cases showed malignant histological features (high cellularity, atypia, necrosis, >4 mitoses/HPF). Yet, statistical analysis showed no correlation between survival and any clinicopathological or molecular parameter - meaning the usual soft-tissue risk stratification models do not reliably predict outcome in primary bone SFTs.
Immunohistochemistry
| Marker | Result | Significance |
|---|---|---|
| STAT6 (nuclear) | Strongly + (100% in bone SFT series) | Most sensitive and specific; pathognomonic |
| CD34 | + in ~95% | Characteristically diffuse and strong |
| CD99 | Often + | Non-specific |
| Bcl-2 | Often + | Non-specific |
| Vimentin | + | Non-specific |
| Factor XIIIa | Variably + | |
| Keratin / EMA | Negative | Helps exclude mesothelioma (which is keratin+) |
| S100 | Negative | |
| SMA / desmin | Usually negative |
The nuclear localisation of STAT6 (visible on IHC) is the key diagnostic discriminator. It reflects the chimeric NAB2::STAT6 protein entering the nucleus. This finding is considered virtually pathognomonic for SFT.
Molecular Confirmation
When IHC is inconclusive:
- RT-PCR for NAB2::STAT6 fusion transcripts - sensitive but requires knowledge of which exons are fused
- Next-generation sequencing (NGS) / RNA sequencing - preferred for comprehensive fusion detection
- FISH - can detect rearrangement at 12q13 but does not identify the specific fusion partner
Diagnosis - Key Differential
| Tumour | How to Distinguish from Bone SFT |
|---|---|
| Ewing sarcoma | EWSR1-FLI1/ERG fusion; CD99 membranous; STAT6 negative |
| Fibrosarcoma | STAT6 negative; CD34 negative; lacks staghorn vessels |
| GCT of bone | Multinucleated giant cells; H3.3 G34W mutation; STAT6 negative |
| Synovial sarcoma | SS18-SSX fusion; TLE1+; biphasic architecture |
| Metastatic HPC/SFT | Primary SFT at another site must be excluded |
| Leiomyosarcoma | SMA+; desmin+; STAT6 negative |
| Low-grade fibromyxoid sarcoma | FUS-CREB3L2 fusion; MUC4+; STAT6 negative |
Treatment
Surgery
- Wide/en-bloc resection with negative margins is the mainstay for localised disease
- Intralesional or marginal resection carries high recurrence risk
- For bone: may involve prosthetic replacement (e.g., proximal femur resection with endoprosthesis, as in reported cases)
- Segmental resection (e.g., mandibulectomy) for jaw SFTs
Radiation
- Adjuvant radiotherapy is recommended for positive or close margins
- Definitive radiotherapy is an option for unresectable lesions and has shown promise for local control
- Fractionated RT is standard when used post-operatively
Systemic Therapy (for advanced/metastatic disease)
- Anthracycline-based chemotherapy (doxorubicin ± ifosfamide) as first-line
- Antiangiogenic agents: pazopanib, sunitinib, sorafenib - increasingly used given the rich vascular nature of SFT; considered after first-line failure
- Imatinib, axitinib, eribulin under investigation
- No established standard for advanced disease; enrolment in clinical trials is recommended
Prognosis
- Pleural/soft tissue SFTs: ~90% behave indolently; ~10% show aggressive course
- Primary bone SFTs behave more aggressively: the Bianchi 2021 series reported disease-specific survival of 64% at 5 years and 49% at 10 years
- Unlike soft-tissue SFTs, histological grade and mitotic count do NOT reliably predict outcome in bone SFTs
- Risk factors for recurrence include incomplete resection and late metastasis (bone, liver, lung are metastatic targets)
- Intracranial SFT/HPC has a 60-80% local recurrence rate and 25% rate of systemic metastasis, predominantly to bone and liver
Key Points Summary
- Primary intraosseous SFT is extremely rare and frequently misdiagnosed (Ewing sarcoma is a common initial misdiagnosis)
- The NAB2::STAT6 fusion on 12q13 is pathognomonic - confirmed by RT-PCR, NGS, or nuclear STAT6 IHC
- CD34+ / STAT6 nuclear+ immunoprofile is diagnostic
- Bone SFTs have a more aggressive clinical course than soft tissue counterparts, and standard soft-tissue grading criteria are not reliably prognostic in bone
- Wide surgical resection with clear margins is the most important determinant of outcome
- Adjuvant/definitive radiotherapy and antiangiogenic agents are useful in unresectable or metastatic settings
- Long-term follow-up (>10 years) is mandatory given late recurrences and metastases
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Solitary Fibrous Tumor
- Murray & Nadel's Textbook of Respiratory Medicine - SOLITARY FIBROUS TUMOR
- Bradley and Daroff's Neurology in Clinical Practice - Hemangiopericytoma and SFT
- Bianchi et al. (2021). Clinical, Histological, and Molecular Features of Solitary Fibrous Tumor of Bone. Cancers. PMID 34069378
- Ren et al. (2024). Advances in the molecular biology of SFT. Cancer Metastasis Rev. PMID 39120790
- Janik et al. (2023). Diagnostics and Treatment of Extrameningeal SFTs. Cancers. PMID 38136399
- Argyris et al. (2024). Primary Intraosseous SFT of the Mandible. Head Neck Pathol. PMID 39621174
- Coppola et al. (2022). Primary intraosseous SFT - case report and review. Pathologica. PMID 36305024
This is a shared conversation. Sign in to Orris to start your own chat.